
Validation: the CamSol solubility score is tested against experimentally determined solubility changes upon mutation from the literature. The testing set contains 56 different protein variants from 19 wild type proteins and includes some antibodies. The bar plot reports the fraction of correctly predicted solubility changes upon mutation using CamSol and two methods of predicting solubility upon over-expression that are SOLpro, and PROSO II. See Reference 1 for details.
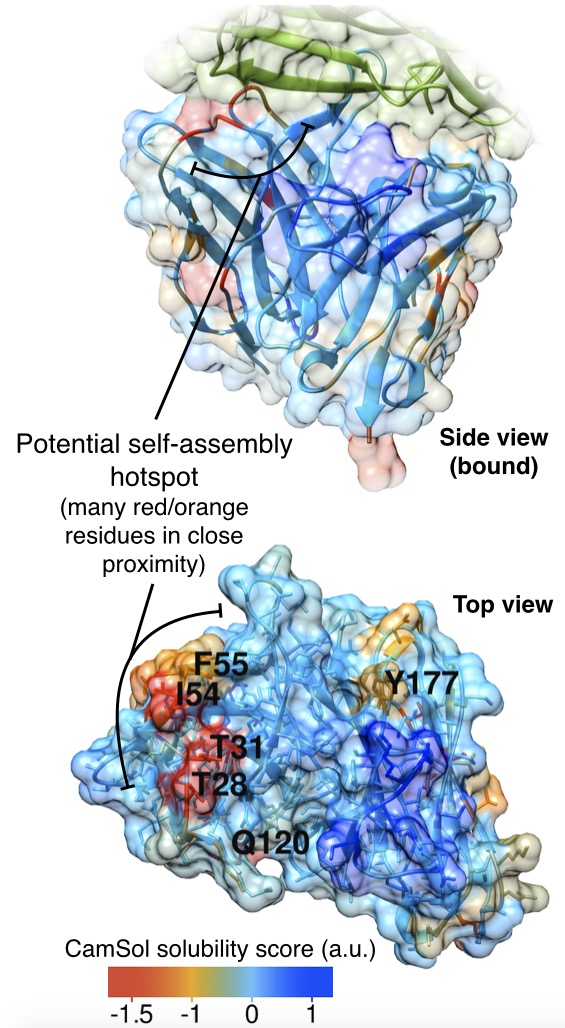
Identification of self-assembly hotspots: The CamSol structurally corrected profile is colour-coded on the surface of a scFv to identify potential self-assembly hotspots, which consist in patches of solvent-exposed poorly soluble amino acids.
The CamSol Method
-
Rational design of protein variants with enhanced solubility.
-
Fast solubility screening of protein libraries.
-
Identification of aggregation-promoting hotspots.

The CamSol method of protein solubility prediction comprises three algorithms that can be used individually for specific tasks or together to rationally design protein variants with enhanced solubility.
These algorithms are:
- A fast sequence-based predictor of intrinsic solubility profiles and solubility scores. The profiles consist in a score for each residue and represent its impact on the overall solubility of the protein molecule under scrutiny, while the solubility score can be used very effectively to rank different protein variants (i.e. protein with some degree of sequence similarity). This algorithm can be used on its own to quickly screen computationally protein libraries for solubility.
- An algorithm that exploits the knowledge of the native structure to perform structural corrections to the intrinsic solubility profile. This accounts for the proximity of the amino acids in the three-dimensional structure and for their solvent exposure. The structurally corrected profile can be color-coded on the structure of the protein to spot patches of low solubility that may elicit the self-assembly process.
- An algorithm that analyses the structurally corrected profile to identify suitable sites for amino acids substitution or insertion. Mutations at these sites are predicted to have a maximum impact on the solubility of the protein while retaining the native structure.
Usage
For use by commercial organisations please contact Professor Michele Vendruscolo.
Academic users can access the CamSol web server at the Vendruscolo Lab software website.
Fast solubility screening of protein libraries
The intrinsic solubility score computed from the amino acid sequence by the first algorithm can be used to rank libraries of protein variants according to their solubility. For example, in vitro antibody discovery techniques (e.g. phage display) usually yield a large number of antibody variants that bind their antigen with high affinity. Since these variants share a high degree of sequence similarity, the CamSol method will produce accurate solubility rankings, reducing the need for experiments and helping the selection of the best candidates for development.
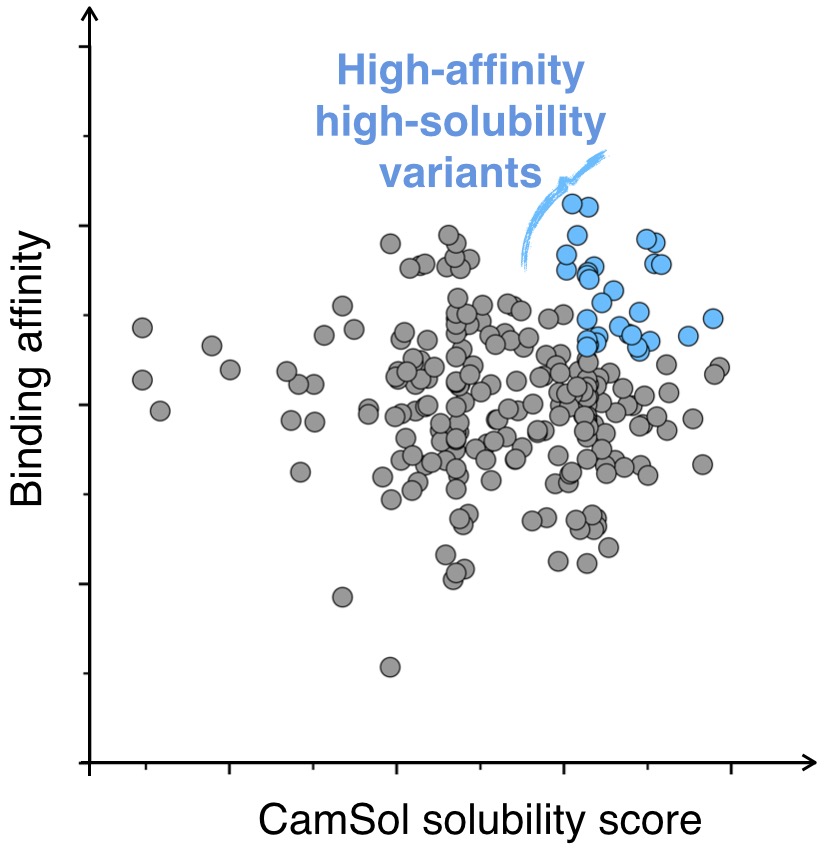
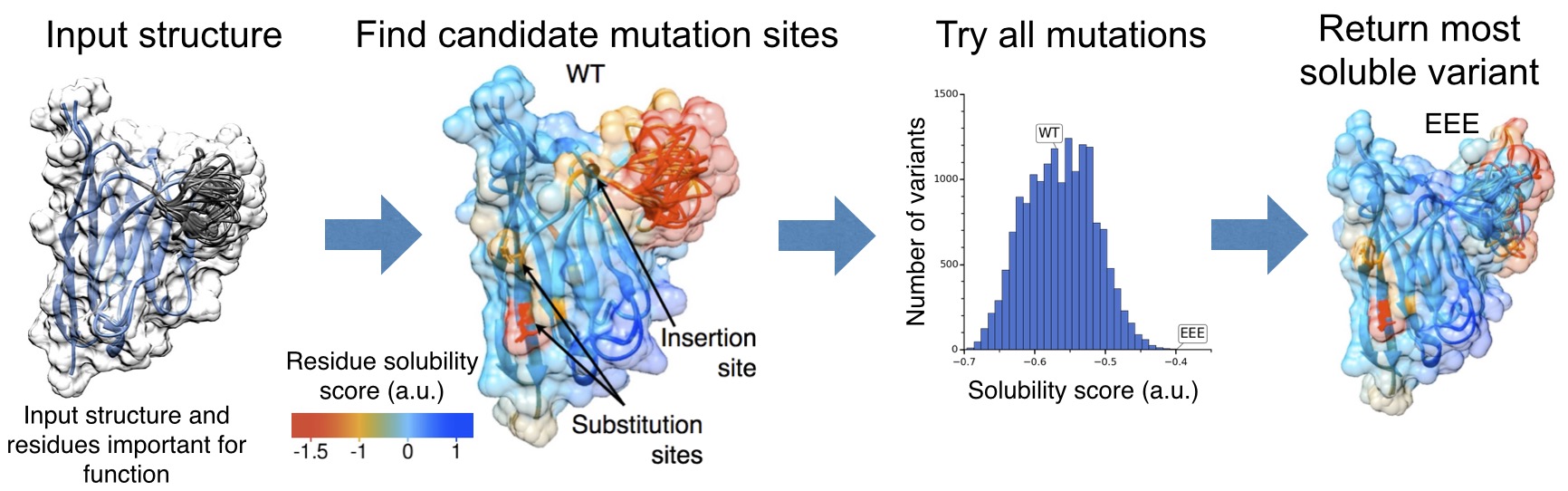
Rational design of protein variants with enhanced solubility
The three algorithms that constitute the CamSol method can be used together for the structure-based design of soluble protein variants. In this embodiment the method performs in silico a rapid and systematic computational screening of tens of thousands of possible amino acid substitutions or insertions to identify specific mutations that are predicted to maximally increase the solubility of a protein while preserving its fundamental properties, including its functional structure and binding affinity. The method requires the knowledge of the native structure of the target protein, which could be available by experimental or by computational (e.g. homology modeling) techniques (high resolution is not required). The structural correction is used to distinguish, among the residues that are classified as poorly soluble, those that are required for functional reasons (e.g. the residues that form the hydrophobic core) from those that remain exposed to the solvent and are not strictly necessary. One can also provide a list of residues important for function or that cannot be otherwise mutated and the maximum number of mutations that the algorithm is allowed to perform so that the wild-type sequence is largely conserved. Four steps are automatically performed by the method: (i) Calculation of the intrinsic solubility profile, (ii) calculation of the structural correction to the intrinsic solubility profile, (iii) identification of suitable mutation sites using the structurally corrected solubility profile, and (iv) screening of all possible mutations/insertions at those site to identify the most soluble variant.
References
P. Sormanni, F. A. Aprile and M. Vendruscolo.
The CamSol method of rational design of protein mutants with enhanced solubility.
J. Mol. Biol. 2015. doi:10.1016/j.jmb.2014.09.026.
P. Sormanni, L. Amery, S. Ekizoglou, M. Vendruscolo, and B. Popovic.
Rapid and accurate in silico solubility screening of a monoclonal antibody library.
Sci. Rep. 2017. doi:10.1038/s41598-017-07800-w.