

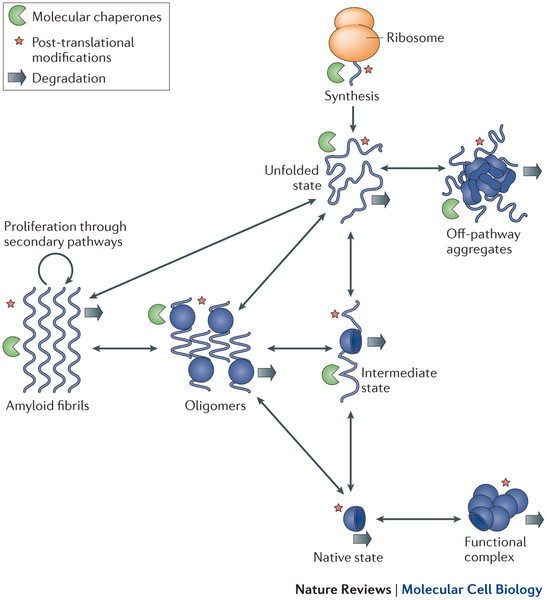


LATEST RESEARCH
Discovery of potent inhibitors of alpha-synuclein aggregation using structure-based iterative learning. Nat. Chem. Biol. (2024)
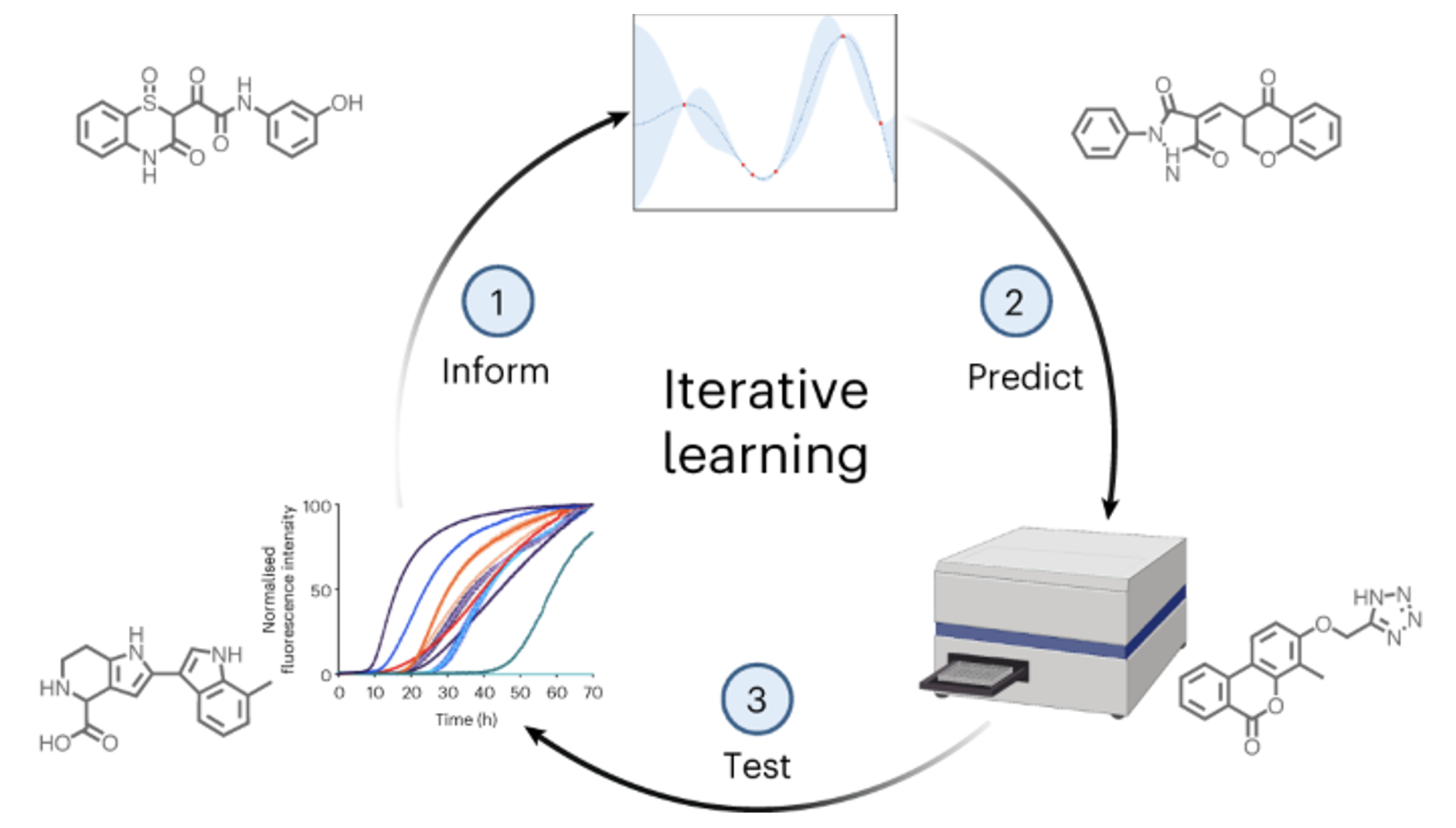
Machine learning methods hold the promise to reduce the costs and the failure rates of conventional drug discovery pipelines. This issue is especially pressing for neurodegenerative diseases, where the development of disease-modifying drugs has been particularly challenging. To address this problem, we describe here a machine learning approach to identify small molecule inhibitors of alpha-synuclein aggregation, a process implicated in Parkinson's disease and other synucleinopathies. Because the proliferation of alpha-synuclein aggregates takes place through autocatalytic secondary nucleation, we aim to identify compounds that bind the catalytic sites on the surface of the aggregates. To achieve this goal, we use structure-based machine learning in an iterative manner to first identify and then progressively optimize secondary nucleation inhibitors. Our results demonstrate that this approach leads to the facile identification of compounds two orders of magnitude more potent than previously reported ones.
Pharmacological inhibition of alpha-synuclein aggregation within liquid condensates. Nat. Comm. (2024)
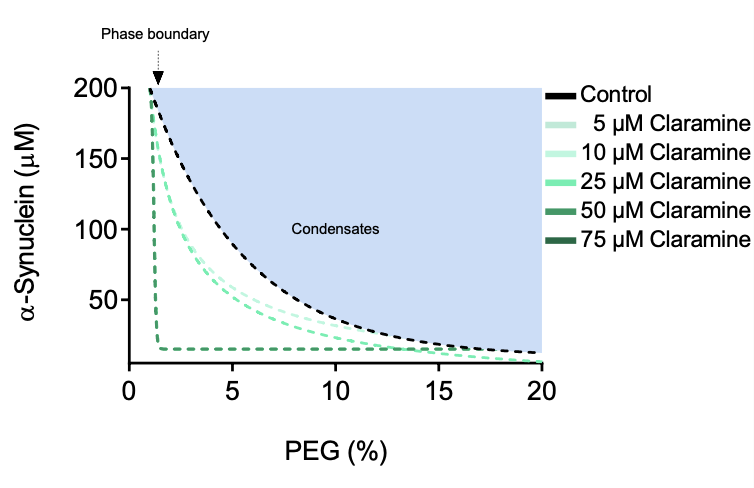
Aggregated forms of alpha-synuclein constitute the major component of Lewy bodies, the proteinaceous aggregates characteristic of Parkinson's disease. Emerging evidence suggests that alpha-synuclein aggregation may occur within liquid condensates formed through phase separation. This mechanism of aggregation creates new challenges and opportunities for drug discovery for Parkinson's disease, which is otherwise still incurable. Here we show that the condensation-driven aggregation pathway of alpha-synuclein can be inhibited using small molecules. We report that the aminosterol claramine stabilizes alpha-synuclein condensates and inhibits alpha-synuclein aggregation within the condensates both in vitro and in a Caenorhabditis elegans model of Parkinson's disease. By using a chemical kinetics approach, we show that the mechanism of action of claramine is to inhibit primary nucleation within the condensates. These results illustrate a possible therapeutic route based on the inhibition of protein aggregation within condensates, a phenomenon likely to be relevant in other neurodegenerative disorders.
Spontaneous nucleation and fast aggregate-dependent proliferation of alpha-synuclein aggregates within liquid condensates at neutral pH.
Proc. Natl. Acad. Sci. USA (2023)
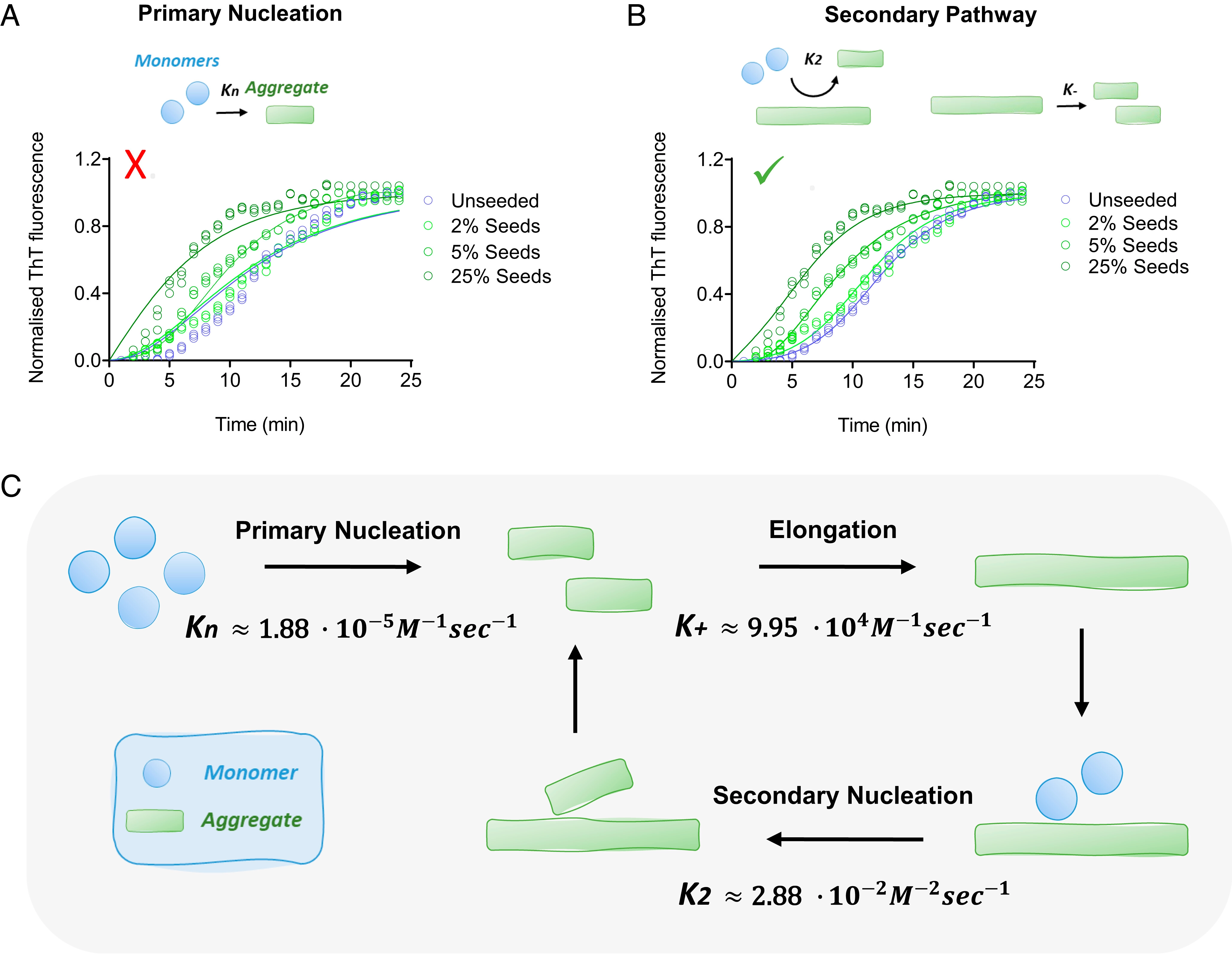
The aggregation of alpha-synuclein into amyloid fibrils has been under scrutiny in recent years because of its association with Parkinson's disease. This process can be triggered by a lipid-dependent nucleation process, and the resulting aggregates can proliferate through secondary nucleation under acidic pH conditions. It has also been recently reported that the aggregation of alpha-synuclein may follow an alternative pathway, which takes place within dense liquid condensates formed through phase separation. The microscopic mechanism of this process, however, remains to be clarified. Here, we used fluorescence-based assays to enable a kinetic analysis of the microscopic steps underlying the aggregation process of alpha-synuclein within liquid condensates. Our analysis shows that at pH 7.4, this process starts with spontaneous primary nucleation followed by rapid aggregate-dependent proliferation. Our results thus reveal the microscopic mechanism of alpha-synuclein aggregation within condensates through the accurate quantification of the kinetic rate constants for the appearance and proliferation of alpha-synuclein aggregates at physiological pH.
Protein condensation diseases: therapeutic opportunities.
Nat. Comm. (2022)
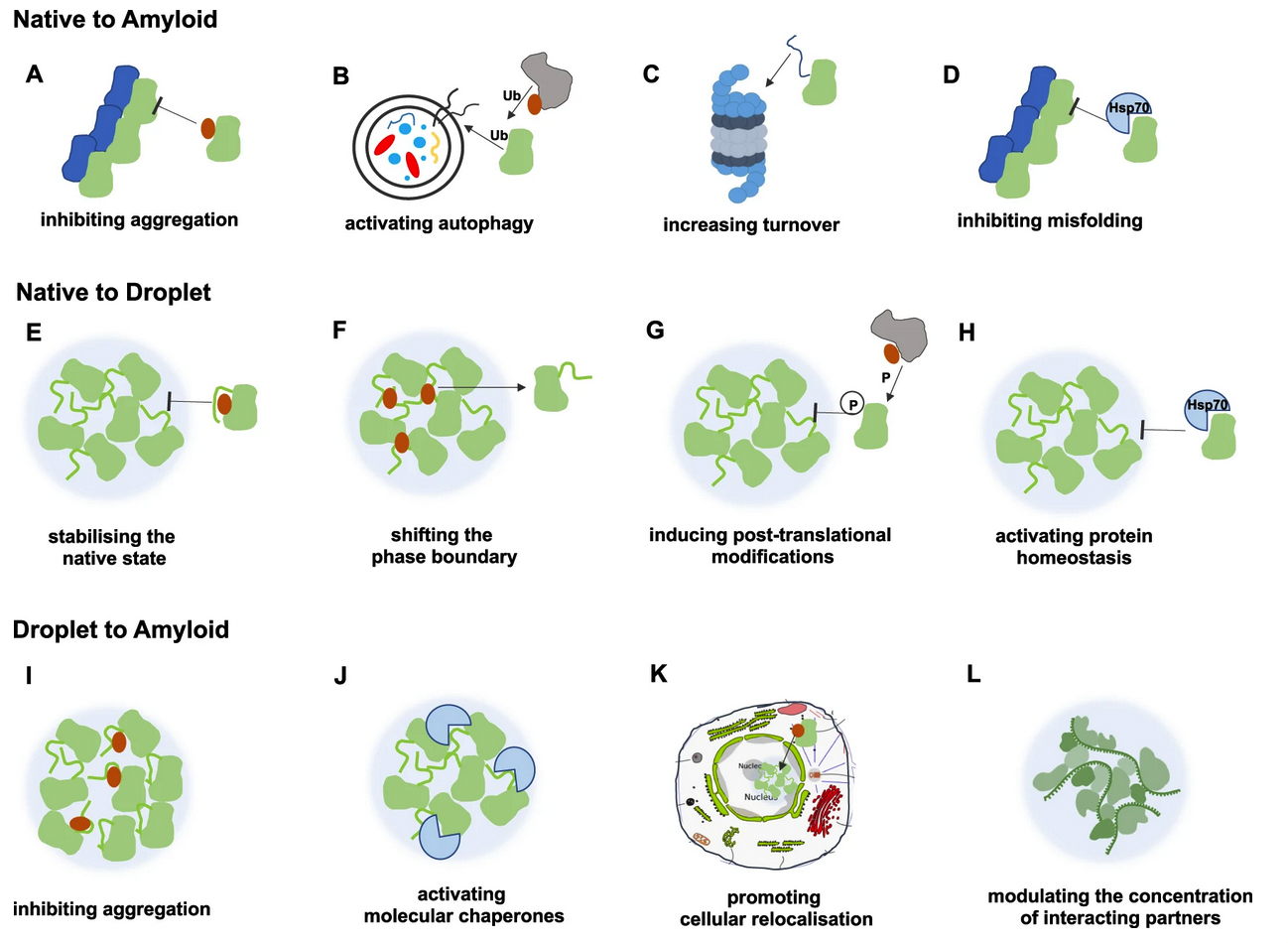
Condensed states of proteins, including liquid-like membraneless organelles and solid-like aggregates, contribute in fundamental ways to the organisation and function of the cell. Perturbations of these states can lead to a variety of diseases through mechanisms that we are now beginning to understand. We define protein condensation diseases as conditions caused by the disruption of the normal behaviour of the condensed states of proteins. We analyze the problem of the identification of targets for pharmacological interventions for these diseases and explore opportunities for the regulation of the formation and organisation of aberrant condensed states of proteins.
Generic nature of the condensed states of proteins. Nat. Cell Biol. (2021)
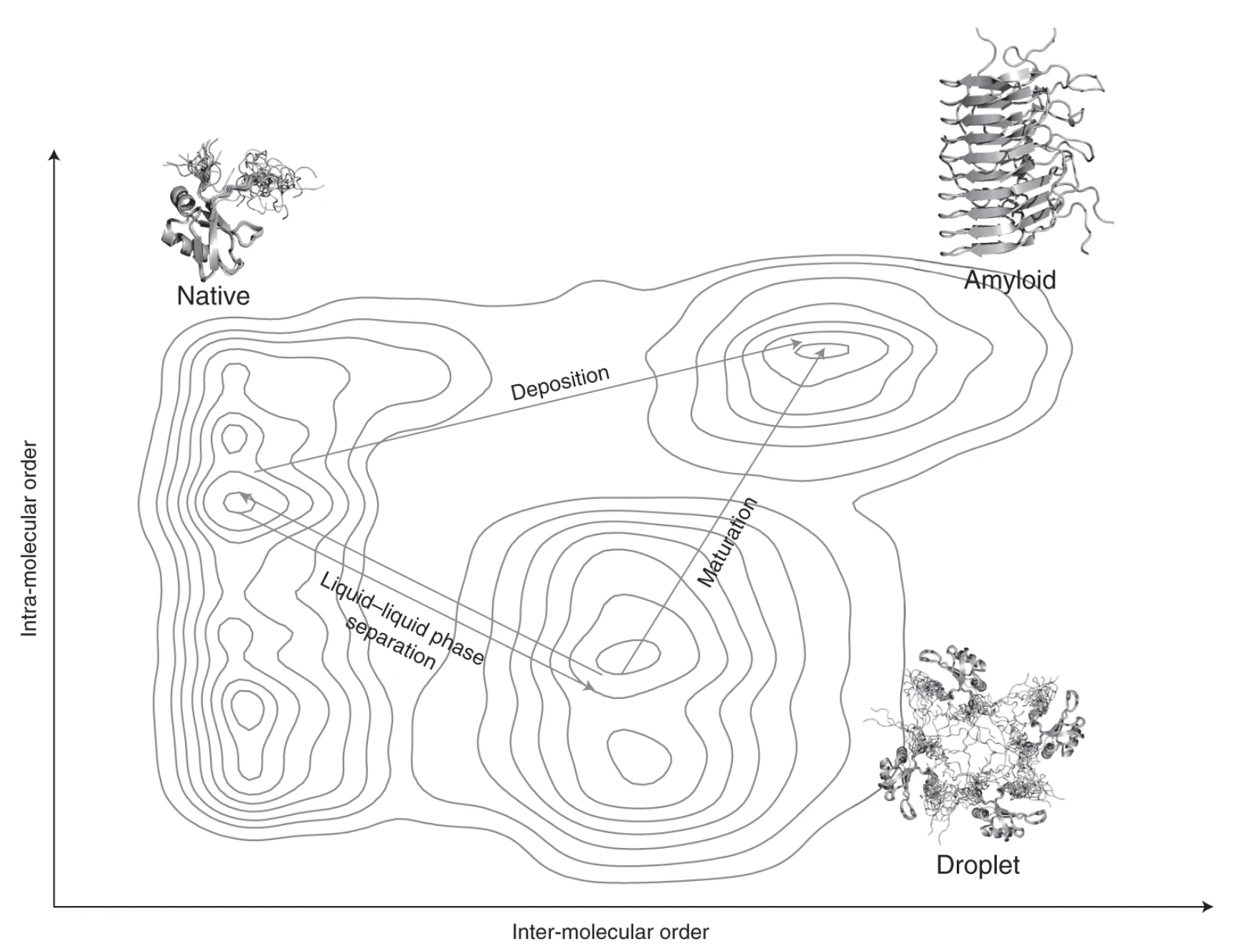
Proteins undergoing liquid-liquid phase separation are being discovered at an increasing rate. Since at the high concentrations present in the cell most proteins would be expected to form a liquid condensed state, this state should be considered to be a fundamental state of proteins along with the native state and the amyloid state. Here we discuss the generic nature of the liquid-like and solid-like condensed states, and describe a wide variety of biological functions conferred by these condensed states.
Systematic activity maturation of a single-domain antibody with non-canonical amino acids through chemical mutagenesis.
Cell Chemical Biology (2021).
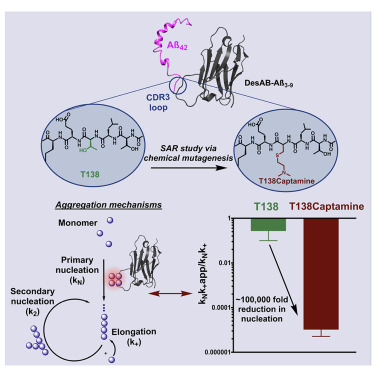
Great advances have been made over the last four decades in therapeutic and diagnostic applications of antibodies. The activity maturation of antibody candidates, however, remains a significant challenge. To address this problem, we present a method that enables the systematic enhancement of the activity of a single-domain antibody through the post-translational installation of non-canonical side chains by chemical mutagenesis. We illustrate this approach by performing a structure-activity relationship study beyond the 20 naturally occurring amino acids on a single-domain antibody designed in silico to inhibit the aggregation of the amyloid-beta peptide, a process closely linked to Alzheimer's disease. We found that this approach can improve, by five orders of magnitude, the anti-aggregation activity of the starting single-domain antibody, without affecting its stability. These results show that the expansion of the chemical space available to antibodies through chemical mutagenesis can be exploited for the systematic enhancement of the activity of these molecules.
Rational design of a conformation-specific antibody for the quantification of Abeta oligomers. Proc. Natl. Acad. Sci USA (2020).
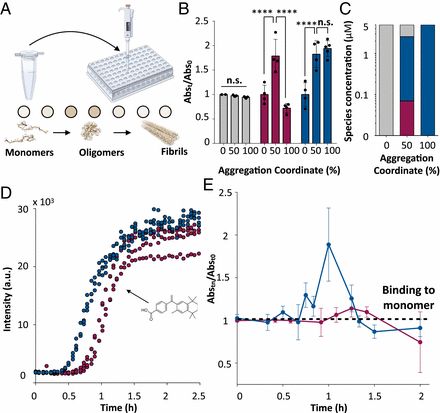
Protein misfolding and aggregation is the hallmark of numerous human disorders, including Alzheimer's disease. This process involves the formation of transient and heterogeneous soluble oligomers, some of which are highly cytotoxic. A major challenge for the development of effective diagnostic and therapeutic tools is thus the detection and quantification of these elusive oligomers. Here, to address this problem, we develop a two-step rational design method for the discovery of oligomer-specific antibodies. The first step consists of an 'antigen scanning' phase in which an initial panel of antibodies is designed to bind different epitopes covering the entire sequence of a target protein. This procedure enables the determination through in vitro assays of the regions exposed in the oligomers but not in the fibrillar deposits. The second step involves an 'epitope mining' phase, in which a second panel of antibodies is designed to specifically target the regions identified during the scanning step. We illustrate this method in the case of the amyloid beta (Abeta) peptide, whose oligomers are associated with Alzheimer's disease. Our results show that this approach enables the accurate detection and quantification of Abeta oligomers in vitro, and in Caenorhabditis elegans and mouse hippocampal tissues.
Proteome-wide observation of the phenomenon of life on the edge of solubility. Proc. Natl. Acad. Sci USA (2020).
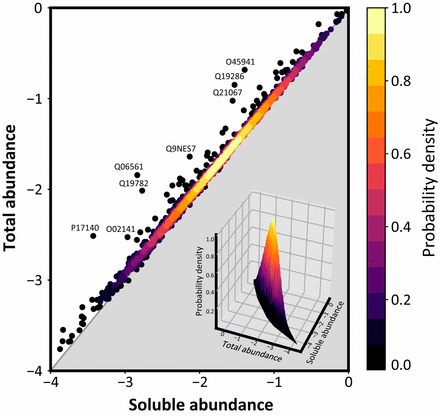
To function effectively proteins must avoid aberrant aggregation, and hence they are expected to be expressed at concentrations safely below their solubility limits. By analyzing proteome-wide mass spectrometry data of Caenorhabditis elegans, however, we show that the levels of about three-quarters of the nearly 4,000 proteins analyzed in adult animals are close to their intrinsic solubility limits, indeed exceeding them by about 10% on average. We next asked how aging and functional self-assembly influence these solubility limits. We found that despite the fact that the total quantity of proteins within the cellular environment remains approximately constant during aging, protein aggregation sharply increases between days 6 and 12 of adulthood, after the worms have reproduced, as individual proteins lose their stoichiometric balances and the cellular machinery that maintains solubility undergoes functional decline. These findings reveal that these proteins are highly prone to undergoing concentration-dependent phase separation, which on aging is rationalized in a decrease of their effective solubilities, in particular for proteins associated with translation, growth, reproduction, and the chaperone system.
Small molecule sequestration of amyloid-beta as a drug discovery strategy for Alzheimer's disease. Sci. Adv. (2020)
Disordered proteins are challenging therapeutic targets, and no drug is currently in clinical use that modifies the properties of their monomeric states. Here, we identify a small molecule (10074-G5) capable of binding and sequestering the intrinsically disordered amyloid-beta (Abeta) peptide in its monomeric, soluble state. Our analysis reveals that this compound interacts with Abeta and inhibits both the primary and secondary nucleation pathways in its aggregation process. We characterize this interaction using biophysical experiments and integrative structural ensemble determination methods. We observe that this molecule increases the conformational entropy of monomeric Abeta while decreasing its hydrophobic surface area. We also show that it rescues a Caenorhabditis elegans model of Abeta-associated toxicity, consistent with the mechanism of action identified from the in silico and in vitro studies. These results illustrate the strategy of stabilizing the monomeric states of disordered proteins with small molecules to alter their behavior for therapeutic purposes.


TOOLS
FuzDrop: Sequence-based prediction of liquid-liquid phase separation propensity of proteins
Web server
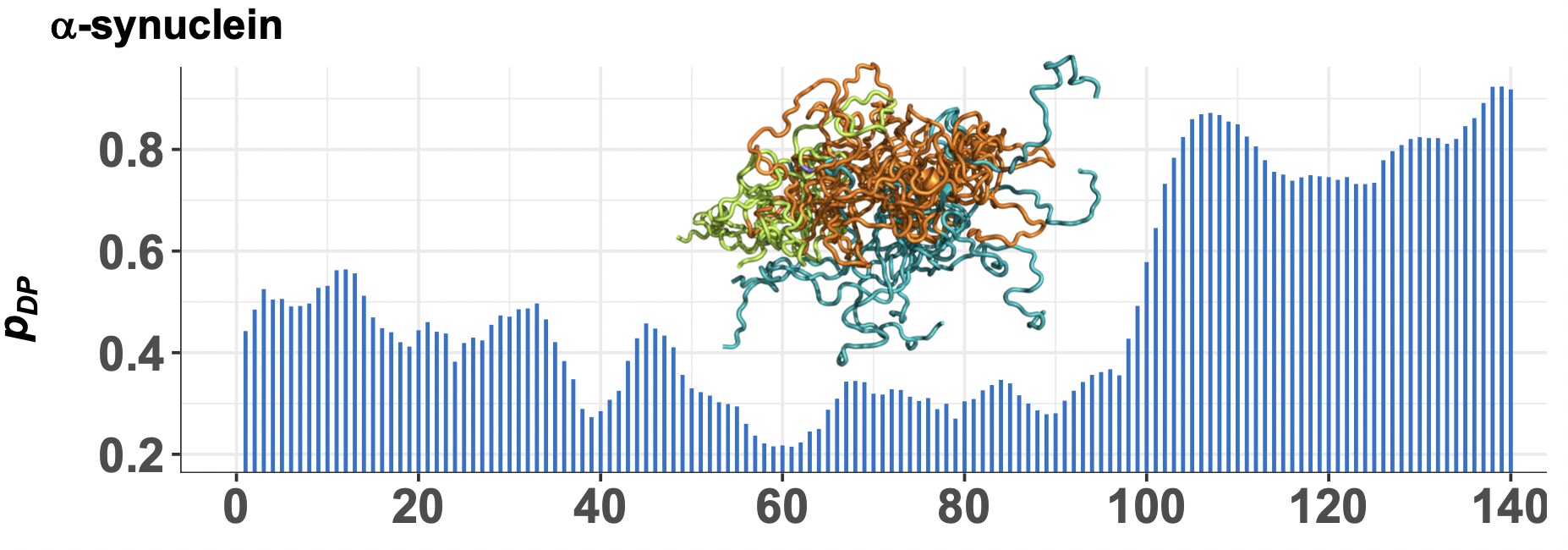
M. C. Hardenberg, A. Horvath, M. Fuxreiter and M. Vendruscolo.
Proc. Natl. Acad. Sci. USA, 117, 33254-33262 (2020).


TOOLS
CamSol:
A method of rational design of protein variants with enhanced solubility.
Web server (academic)
Web server (non academic)
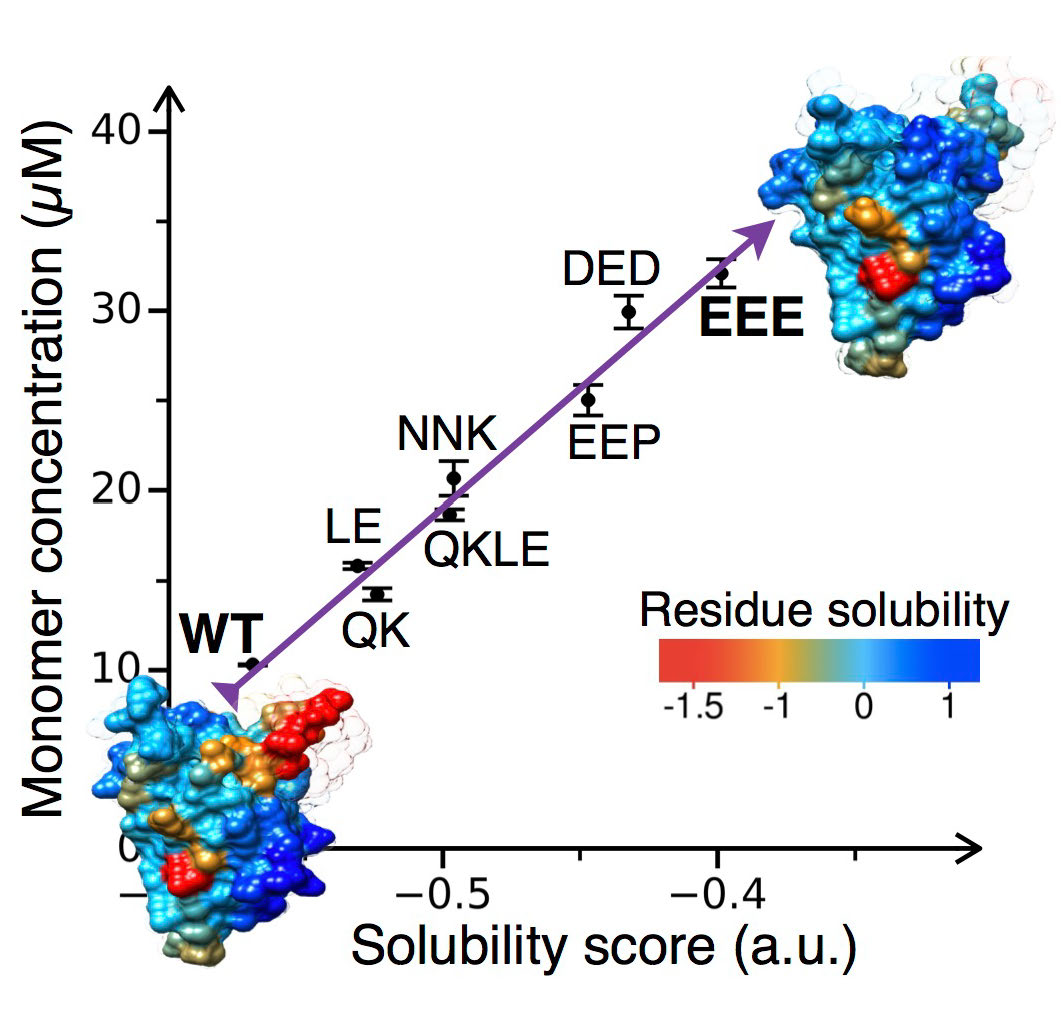
P. Sormanni, F. A. Aprile and M. Vendruscolo.
J. Mol. Biol. 427, 478-490 (2015).
TOOLS
The s2D method: Simultaneous sequence-based prediction of the statistical populations of ordered and disordered regions in proteins
Web server
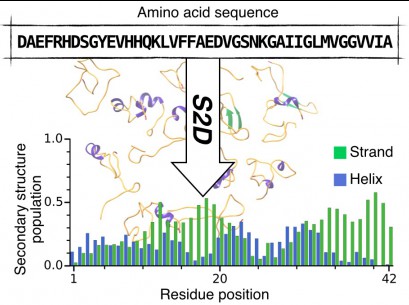
P. Sormanni, C. Camilloni, P. Fariselli and M. Vendruscolo.
J. Mol. Biol. 427, 982-996 (2015).