Discovery of potent inhibitors of alpha-synuclein aggregation using structure-based iterative learning. Nat. Chem. Biol. (2024)
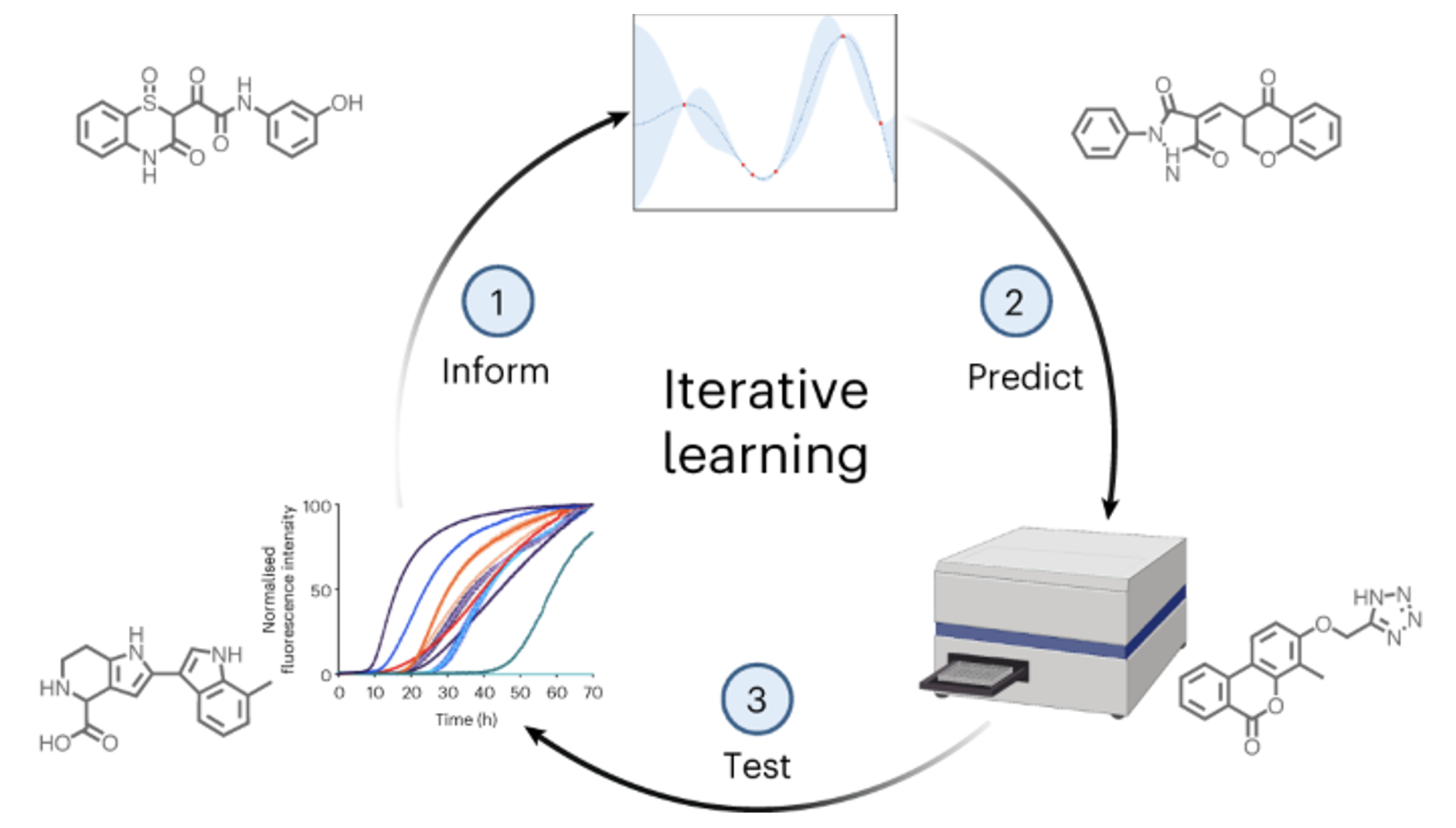
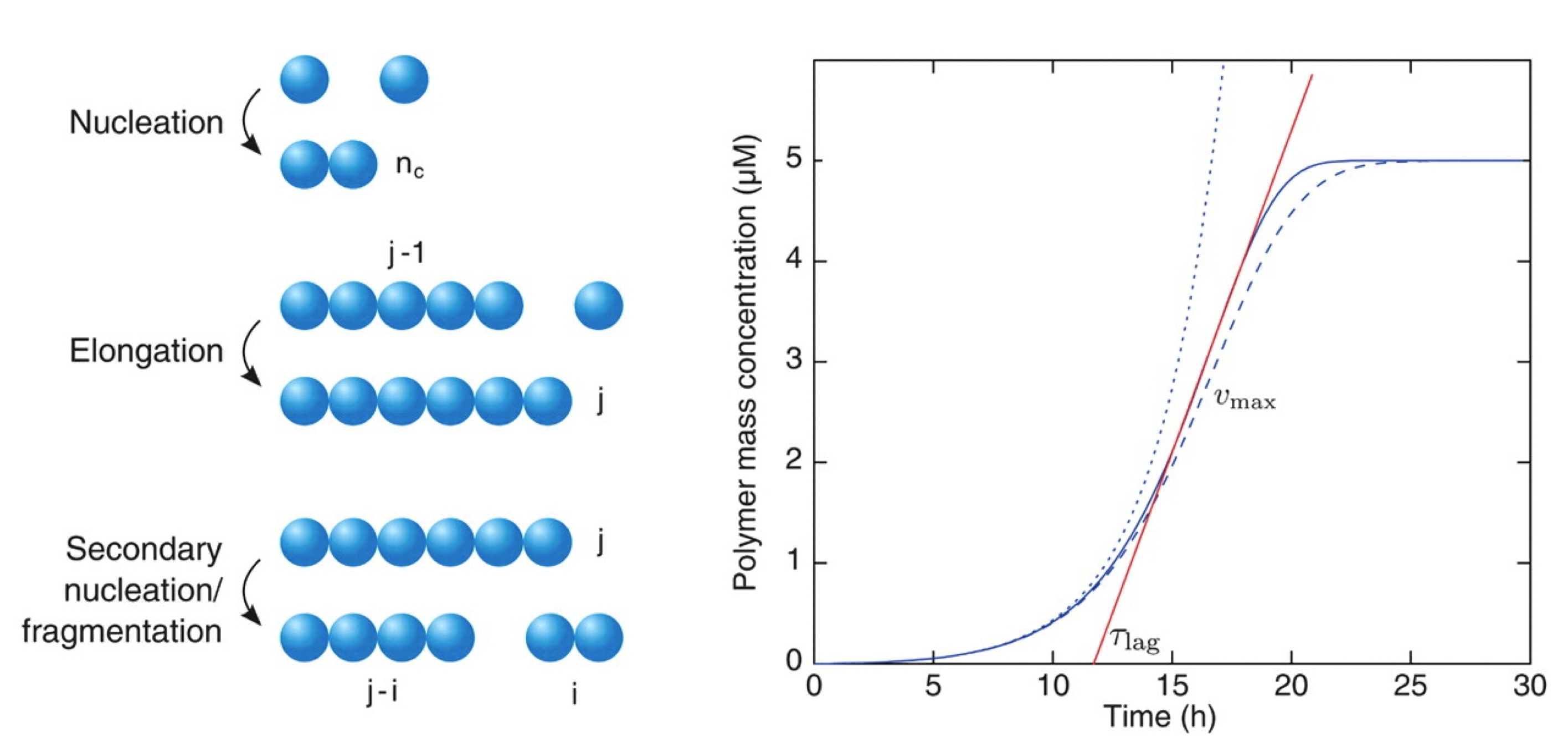
Machine learning methods hold the promise to reduce the costs and the failure rates of conventional drug discovery pipelines. This issue is especially pressing for neurodegenerative diseases, where the development of disease-modifying drugs has been particularly challenging. To address this problem, we describe here a machine learning approach to identify small molecule inhibitors of alpha-synuclein aggregation, a process implicated in Parkinson's disease and other synucleinopathies. Because the proliferation of alpha-synuclein aggregates takes place through autocatalytic secondary nucleation, we aim to identify compounds that bind the catalytic sites on the surface of the aggregates. To achieve this goal, we use structure-based machine learning in an iterative manner to first identify and then progressively optimize secondary nucleation inhibitors. Our results demonstrate that this approach leads to the facile identification of compounds two orders of magnitude more potent than previously reported ones.
Pharmacological inhibition of alpha-synuclein aggregation within liquid condensates. Nat. Comm. (2024)
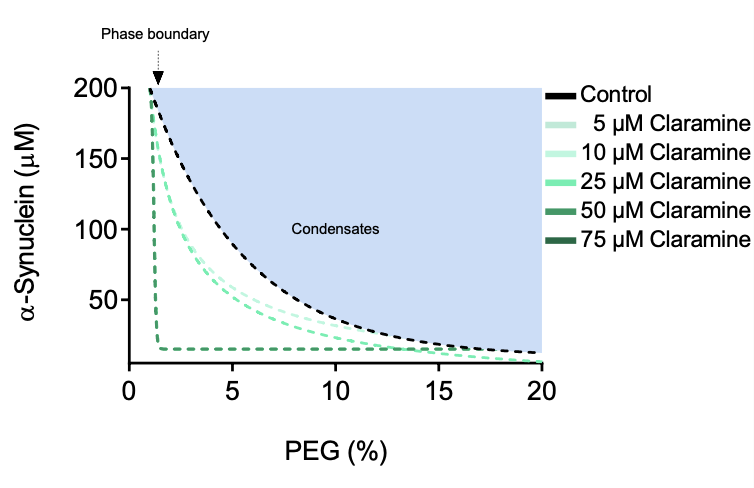
Aggregated forms of alpha-synuclein constitute the major component of Lewy bodies, the proteinaceous aggregates characteristic of Parkinson's disease. Emerging evidence suggests that alpha-synuclein aggregation may occur within liquid condensates formed through phase separation. This mechanism of aggregation creates new challenges and opportunities for drug discovery for Parkinson's disease, which is otherwise still incurable. Here we show that the condensation-driven aggregation pathway of alpha-synuclein can be inhibited using small molecules. We report that the aminosterol claramine stabilizes alpha-synuclein condensates and inhibits alpha-synuclein aggregation within the condensates both in vitro and in a Caenorhabditis elegans model of Parkinson's disease. By using a chemical kinetics approach, we show that the mechanism of action of claramine is to inhibit primary nucleation within the condensates. These results illustrate a possible therapeutic route based on the inhibition of protein aggregation within condensates, a phenomenon likely to be relevant in other neurodegenerative disorders.
Spontaneous nucleation and fast aggregate-dependent proliferation of alpha-synuclein aggregates within liquid condensates at neutral pH.
Proc. Natl. Acad. Sci. USA (2023)
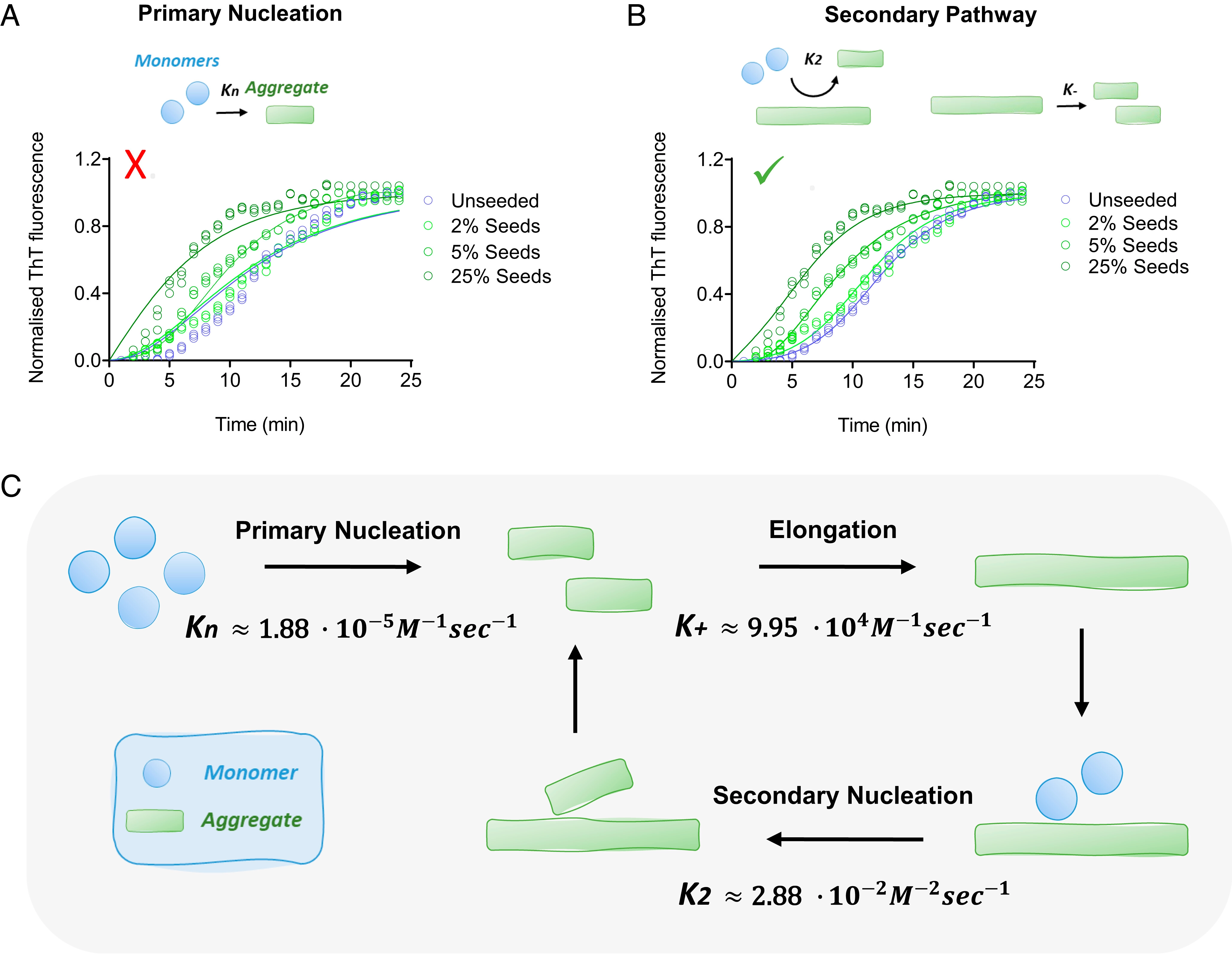
The aggregation of alpha-synuclein into amyloid fibrils has been under scrutiny in recent years because of its association with Parkinson's disease. This process can be triggered by a lipid-dependent nucleation process, and the resulting aggregates can proliferate through secondary nucleation under acidic pH conditions. It has also been recently reported that the aggregation of alpha-synuclein may follow an alternative pathway, which takes place within dense liquid condensates formed through phase separation. The microscopic mechanism of this process, however, remains to be clarified. Here, we used fluorescence-based assays to enable a kinetic analysis of the microscopic steps underlying the aggregation process of alpha-synuclein within liquid condensates. Our analysis shows that at pH 7.4, this process starts with spontaneous primary nucleation followed by rapid aggregate-dependent proliferation. Our results thus reveal the microscopic mechanism of alpha-synuclein aggregation within condensates through the accurate quantification of the kinetic rate constants for the appearance and proliferation of alpha-synuclein aggregates at physiological pH.
Protein condensation diseases: therapeutic opportunities. Nat. Comm. (2022)
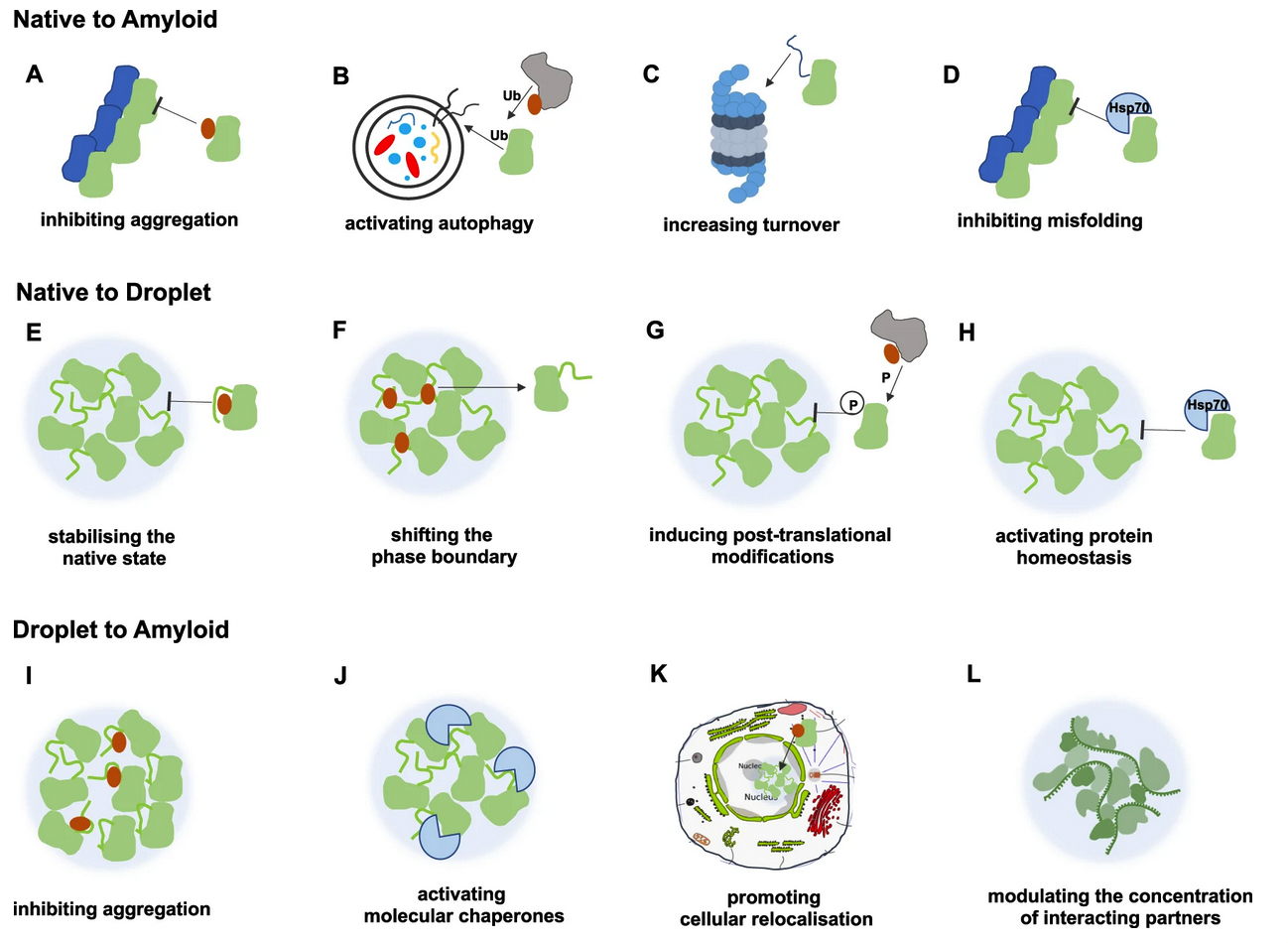
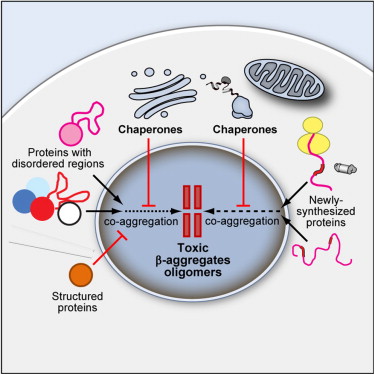
Condensed states of proteins, including liquid-like membraneless organelles and solid-like aggregates, contribute in fundamental ways to the organisation and function of the cell. Perturbations of these states can lead to a variety of diseases through mechanisms that we are now beginning to understand. We define protein condensation diseases as conditions caused by the disruption of the normal behaviour of the condensed states of proteins. We analyze the problem of the identification of targets for pharmacological interventions for these diseases and explore opportunities for the regulation of the formation and organisation of aberrant condensed states of proteins.
Generic nature of the condensed states of proteins. Nat. Cell Biol. (2021)
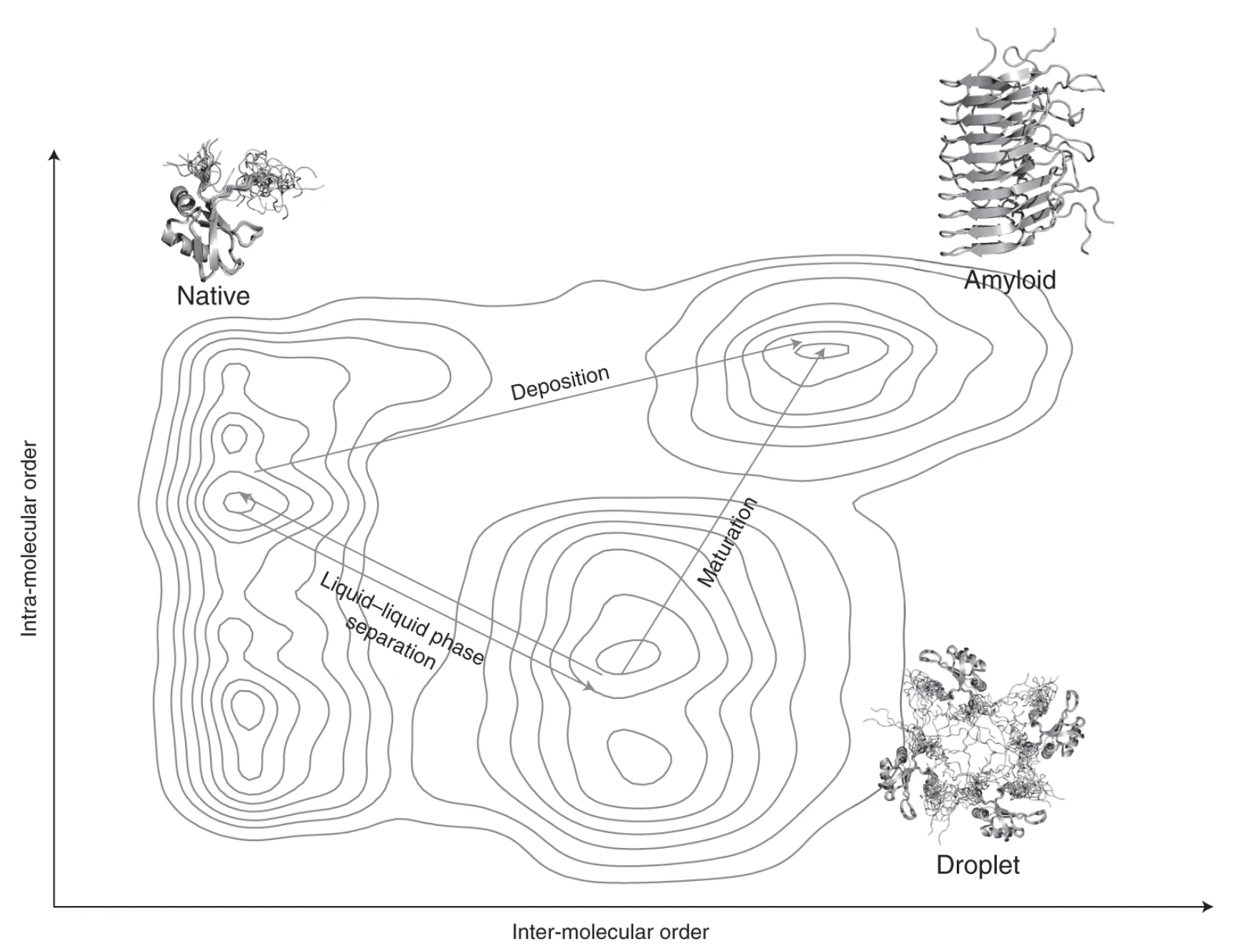
Proteins undergoing liquid-liquid phase separation are being discovered at an increasing rate. Since at the high concentrations present in the cell most proteins would be expected to form a liquid condensed state, this state should be considered to be a fundamental state of proteins along with the native state and the amyloid state. Here we discuss the generic nature of the liquid-like and solid-like condensed states, and describe a wide variety of biological functions conferred by these condensed states.
Systematic activity maturation of a single-domain antibody with non-canonical amino acids through chemical mutagenesis. Cell Chemical Biology (2021).
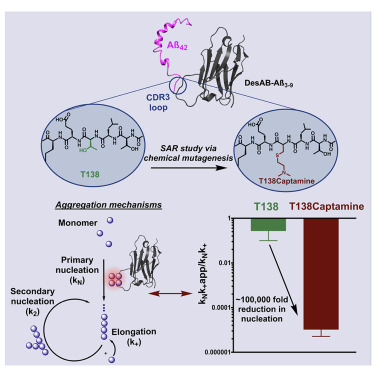
Great advances have been made over the last four decades in therapeutic and diagnostic applications of antibodies. The activity maturation of antibody candidates, however, remains a significant challenge. To address this problem, we present a method that enables the systematic enhancement of the activity of a single-domain antibody through the post-translational installation of non-canonical side chains by chemical mutagenesis. We illustrate this approach by performing a structure-activity relationship study beyond the 20 naturally occurring amino acids on a single-domain antibody designed in silico to inhibit the aggregation of the amyloid-beta peptide, a process closely linked to Alzheimer's disease. We found that this approach can improve, by five orders of magnitude, the anti-aggregation activity of the starting single-domain antibody, without affecting its stability. These results show that the expansion of the chemical space available to antibodies through chemical mutagenesis can be exploited for the systematic enhancement of the activity of these molecules.
Rational design of a conformation-specific antibody for the quantification of Abeta oligomers. Proc. Natl. Acad. Sci USA (2020).
Protein misfolding and aggregation is the hallmark of numerous human disorders, including Alzheimer's disease. This process involves the formation of transient and heterogeneous soluble oligomers, some of which are highly cytotoxic. A major challenge for the development of effective diagnostic and therapeutic tools is thus the detection and quantification of these elusive oligomers. Here, to address this problem, we develop a two-step rational design method for the discovery of oligomer-specific antibodies. The first step consists of an 'antigen scanning' phase in which an initial panel of antibodies is designed to bind different epitopes covering the entire sequence of a target protein. This procedure enables the determination through in vitro assays of the regions exposed in the oligomers but not in the fibrillar deposits. The second step involves an 'epitope mining' phase, in which a second panel of antibodies is designed to specifically target the regions identified during the scanning step. We illustrate this method in the case of the amyloid beta (Abeta) peptide, whose oligomers are associated with Alzheimer's disease. Our results show that this approach enables the accurate detection and quantification of Abeta oligomers in vitro, and in Caenorhabditis elegans and mouse hippocampal tissues.
Proteome-wide observation of the phenomenon of life on the edge of solubility. Proc. Natl. Acad. Sci USA (2020).
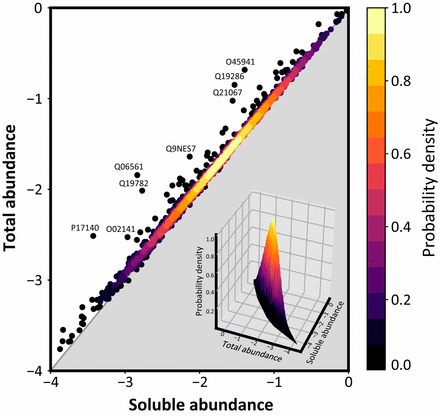
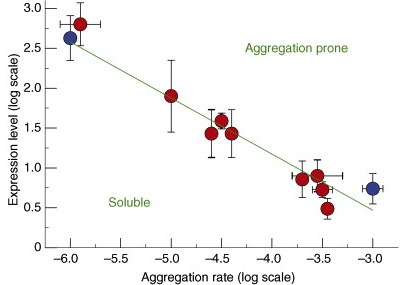
To function effectively proteins must avoid aberrant aggregation, and hence they are expected to be expressed at concentrations safely below their solubility limits. By analyzing proteome-wide mass spectrometry data of Caenorhabditis elegans, however, we show that the levels of about three-quarters of the nearly 4,000 proteins analyzed in adult animals are close to their intrinsic solubility limits, indeed exceeding them by about 10% on average. We next asked how aging and functional self-assembly influence these solubility limits. We found that despite the fact that the total quantity of proteins within the cellular environment remains approximately constant during aging, protein aggregation sharply increases between days 6 and 12 of adulthood, after the worms have reproduced, as individual proteins lose their stoichiometric balances and the cellular machinery that maintains solubility undergoes functional decline. These findings reveal that these proteins are highly prone to undergoing concentration-dependent phase separation, which on aging is rationalized in a decrease of their effective solubilities, in particular for proteins associated with translation, growth, reproduction, and the chaperone system.
Small molecule sequestration of amyloid-beta as a drug discovery strategy for Alzheimer's disease. Sci. Adv. (2020)
Disordered proteins are challenging therapeutic targets, and no drug is currently in clinical use that modifies the properties of their monomeric states. Here, we identify a small molecule (10074-G5) capable of binding and sequestering the intrinsically disordered amyloid-beta (Abeta) peptide in its monomeric, soluble state. Our analysis reveals that this compound interacts with Abeta and inhibits both the primary and secondary nucleation pathways in its aggregation process. We characterize this interaction using biophysical experiments and integrative structural ensemble determination methods. We observe that this molecule increases the conformational entropy of monomeric Abeta while decreasing its hydrophobic surface area. We also show that it rescues a Caenorhabditis elegans model of Abeta-associated toxicity, consistent with the mechanism of action identified from the in silico and in vitro studies. These results illustrate the strategy of stabilizing the monomeric states of disordered proteins with small molecules to alter their behavior for therapeutic purposes.
Supersaturated proteins are enriched at synapses and underlie cell and tissue vulnerability in Alzheimer's disease. Heliyon (2019).
Neurodegenerative disorders progress across the brain in characteristic spatio-temporal patterns. A better understanding of the factors underlying the specific cell and tissue vulnerability responsible for such patterns could help identify the molecular origins of these conditions. To investigate these factors, based on the observation that neurodegenerative disorders are closely associated with the presence of aberrant protein deposits, we made the hypothesis that the vulnerability of cells and tissues is associated to the overall levels of supersaturated proteins, which are those most metastable against aggregation. By analyzing single-cell transcriptomic and subcellular proteomics data on healthy brains of ages much younger than those typical of disease onset, we found that the most supersaturated proteins are enriched in cells and tissues that succumb first to neurodegeneration. Then, by focusing the analysis on a metastable subproteome specific to Alzheimer's disease, we show that it is possible to recapitulate the pattern of disease progression using data from healthy brains. We found that this metastable subproteome is significantly enriched for synaptic processes and mitochondrial energy metabolism, thus rendering the synaptic environment dangerous for aggregation. The present identification of protein supersaturation as a signature of cell and tissue vulnerability in neurodegenerative disorders could facilitate the search for effective treatments by providing clearer points of intervention.
SAR by kinetics for drug discovery for protein misfolding diseases.
Proc. Natl. Acad. Sci USA (2018).
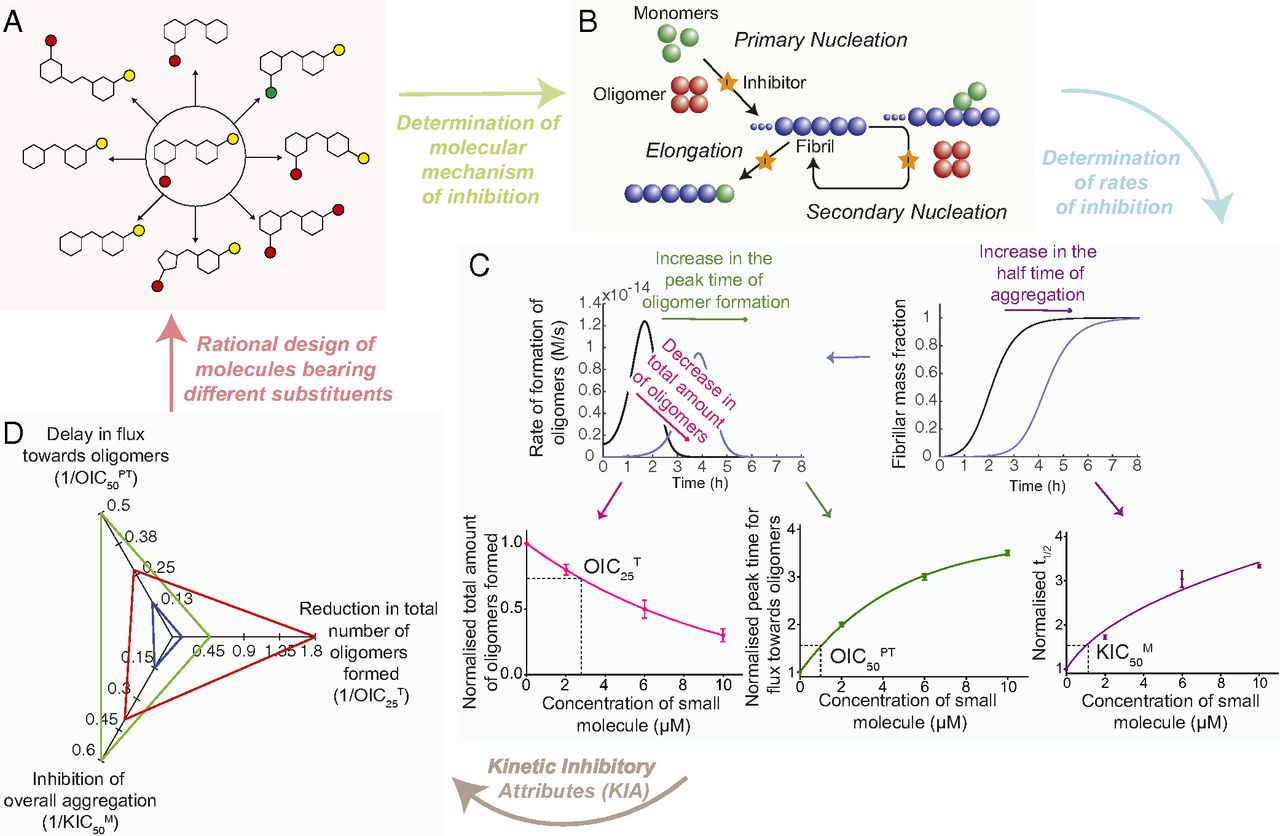
Protein oligomers are increasingly recognized as the most cytotoxic forms of protein aggregates. It has been very challenging, however, to target these oligomers with therapeutic compounds, because of their dynamic and transient nature. To overcome this problem, we present here a 'structure kinetic-activity relationship' (SKAR) approach, which enables the discovery and systematic optimization of compounds that reduce the number of oligomers produced during an aggregation reaction. We illustrate this strategy for the amyloid beta peptide (Abeta), which is closely associated with Alzheimer's disease, by developing a rhodanine compound capable of dramatically reducing the production of Abeta oligomers. As this strategy is general, it can be applied to oligomers of any protein.
Cholesterol catalyses Abeta42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes.
Nature Chemistry (2018).
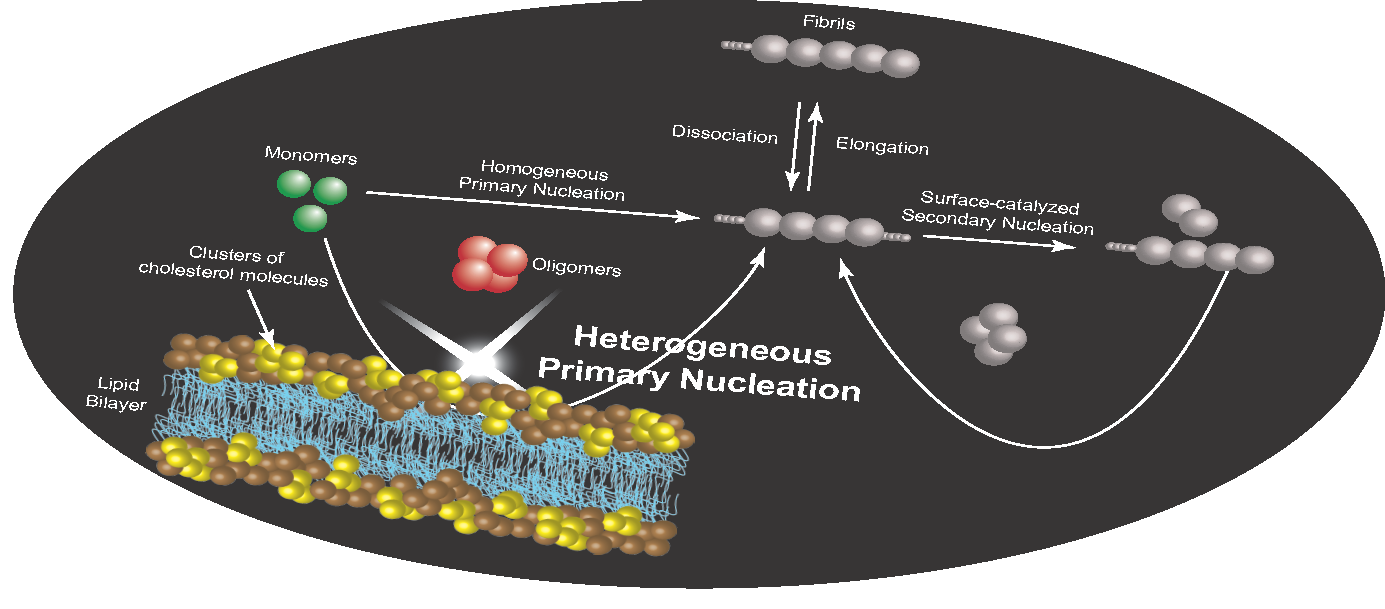
Alzheimer's disease is a neurodegenerative disorder associated with the aberrant aggregation of the amyloid beta peptide. Although increasing evidence implicates cholesterol in the pathogenesis of Alzheimer's disease, the detailed mechanistic link between this lipid molecule and the disease process remains to be fully established. To address this problem, we adopt a kinetics-based strategy that reveals a specific catalytic role of cholesterol in the aggregation of Abeta42 (the 42-residue form of the amyloid beta peptide). More specifically, we demonstrate that lipid membranes containing cholesterol promote Abeta42 aggregation by enhancing its primary nucleation rate by up to 20-fold through a heterogeneous nucleation pathway. We further show that this process occurs as a result of cooperativity in the interaction of multiple cholesterol molecules with Abeta42. These results identify a specific microscopic pathway by which cholesterol dramatically enhances the onset of Abeta42 aggregation, thereby helping rationalize the link between Alzheimer's disease and the impairment of cholesterol homeostasis.
Simultaneous determination of protein structure and dynamics using cryo-electron microscopy. Biophysical Journal (2018).
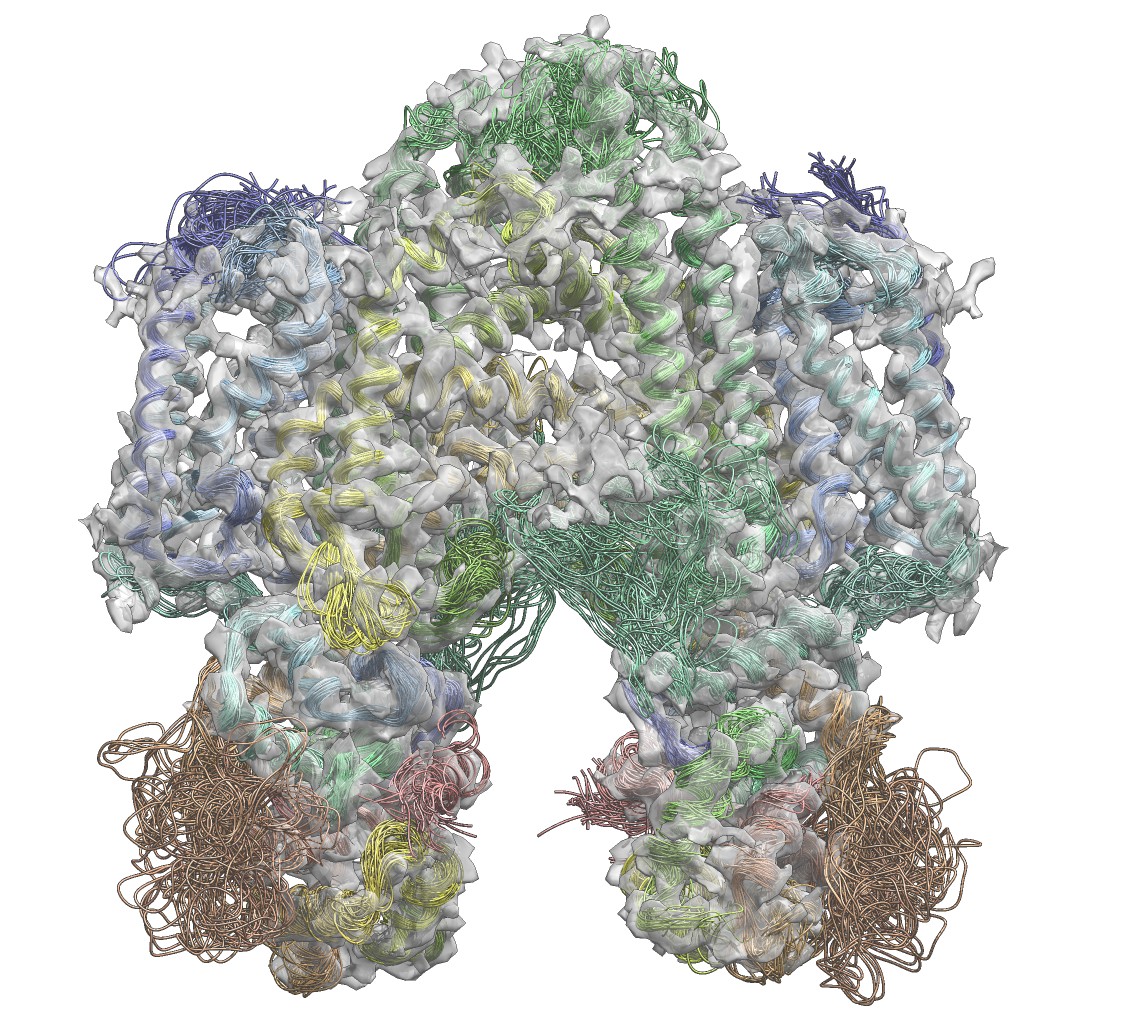
Cryo-electron microscopy is rapidly emerging as a powerful technique to determine the structures of complex macromolecular systems elusive to other techniques. Since many of these systems are highly dynamical, characterising also their movements is a crucial step to unravel their biological functions. To achieve this goal, we report an integrative modelling approach to simultaneously determine structure and dynamics from cryo-electron microscopy density maps. By quantifying the level of noise in the data and dealing with their ensemble-averaged nature, this approach enables the integration of multiple sources of information to model ensembles of structures and infer their populations. We illustrate the method by characterising structure and dynamics of the integral membrane receptor STRA6, thus providing insights into the mechanisms by which it interacts with retinol binding protein and translocates retinol across the membrane.
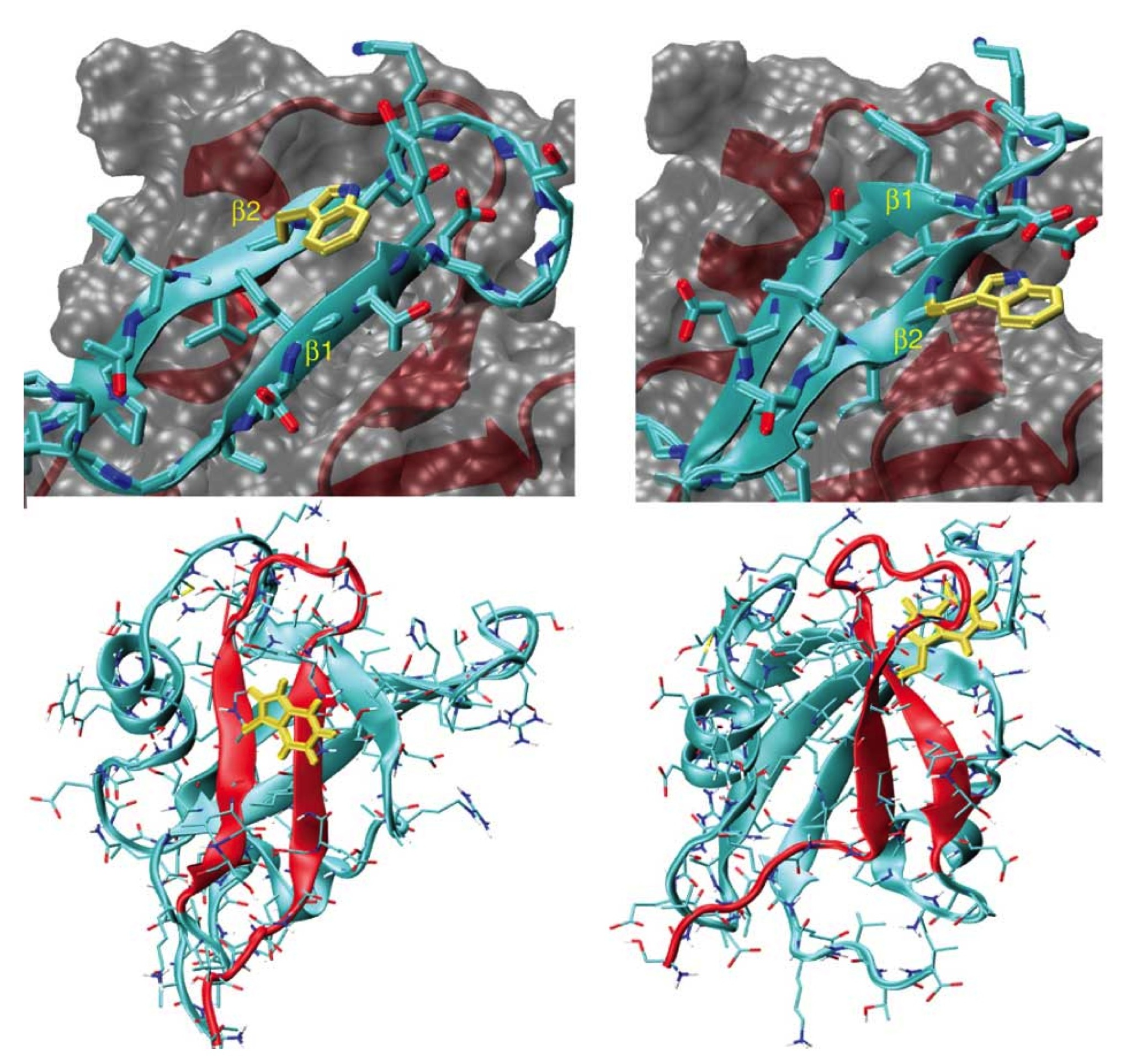
Selective targeting of primary and secondary nucleation pathways in Abeta42 aggregation using a rational antibody scanning method. Science Advances (2017).
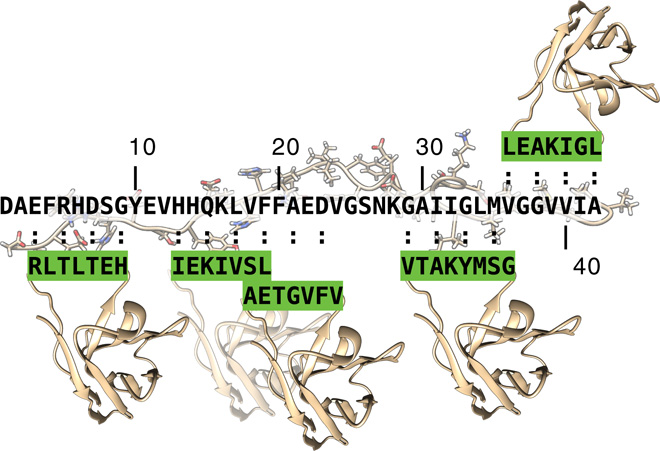
Antibodies targeting Abeta42 are under intense scrutiny because of their therapeutic potential for Alzheimer's disease. To enable systematic searches, we present an “antibody scanning” strategy for the generation of a panel of antibodies against Abeta42. Each antibody in the panel is rationally designed to target a specific linear epitope, with the selected epitopes scanning the Abeta42 sequence. By screening in vitro the panel to identify the specific microscopic steps in the Abeta42 aggregation process influenced by each antibody, we identify two antibodies that target specifically the primary and the secondary nucleation steps, which are key for the production of Abeta42 oligomers. These two antibodies act, respectively, to delay the onset of aggregation and to block the proliferation of aggregates, and correspondingly reduce the toxicity in a Caenorhabditis elegans model overexpressing Abeta42. These results illustrate how the antibody scanning method described here can be used to readily obtain very small antibody libraries with extensive coverage of the sequences of target proteins.
Systematic development of small molecules to inhibit specific microscopic steps of Abeta42 aggregation in Alzheimer's disease. Proc. Natl. Acad. Sci. USA (2017).
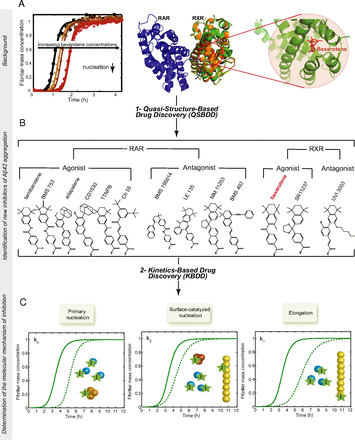
The aggregation of the 42-residue form of the amyloid-beta peptide (Abeta42) is a pivotal event in Alzheimer's disease (AD). The use of chemical kinetics has recently enabled highly accurate quantifications of the effects of small molecules on specific microscopic steps in Abeta42 aggregation. Here, we exploit this approach to develop a rational drug discovery strategy against Abeta42 aggregation that uses as a read-out the changes in the nucleation and elongation rate constants caused by candidate small molecules. We thus identify a pool of compounds that target specific microscopic steps in Abeta42 aggregation. We then test further these small molecules in human cerebrospinal fluid and in a Caenorhabditis elegans model of AD. Our results show that this strategy represents a powerful approach to identify systematically small molecule lead compounds, thus offering an appealing opportunity to reduce the attrition problem in drug discovery.
A protein homeostasis signature in healthy brains recapitulates tissue vulnerability to Alzheimer's disease. Science Advances (2016).
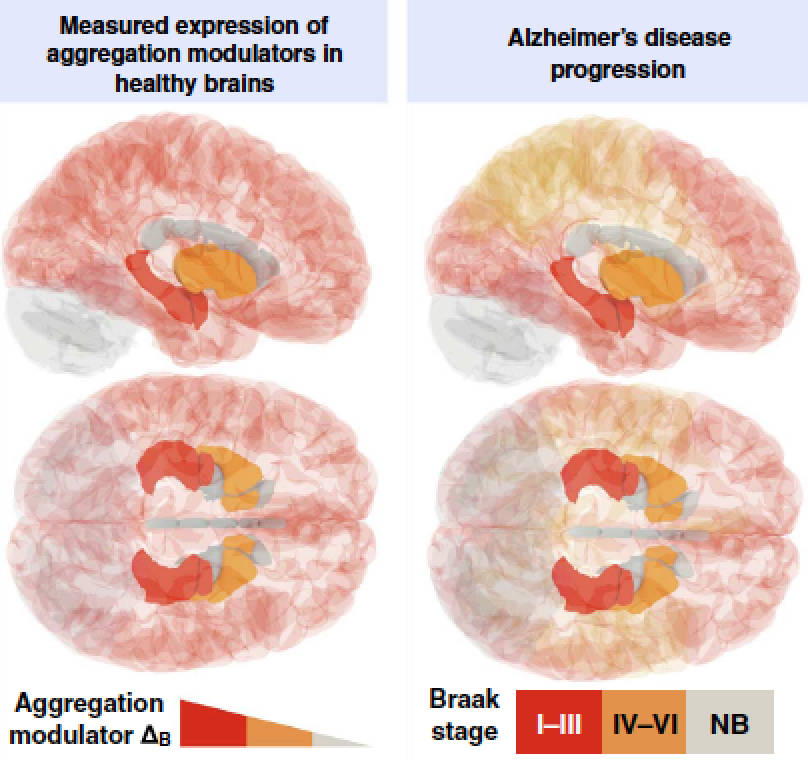
In Alzheimer's disease, aggregates of Abeta and tau in amyloid plaques and neurofibrillary tangles spread progressively across brain tissues following a characteristic pattern, implying a tissue-specific vulnerability to the disease. We report a transcriptional analysis of healthy brains and identify an expression signature that predicts - at ages well before the typical onset - the tissue-specific progression of the disease. We obtain this result by finding a quantitative correlation between the histopathological staging of the disease and the expression patterns of the proteins that coaggregate in amyloid plaques and neurofibrillary tangles, together with those of the protein homeostasis components that regulate Abeta and tau. Because this expression signature is evident in healthy brains, our analysis provides an explanatory link between a tissue-specific environmental risk of protein aggregation and a corresponding vulnerability to Alzheimer's disease.
An anti-cancer drug suppresses the primary nucleation reaction that initiates the formation of toxic Abeta aggregates linked with Alzheimer's disease. Science Advances (2016).
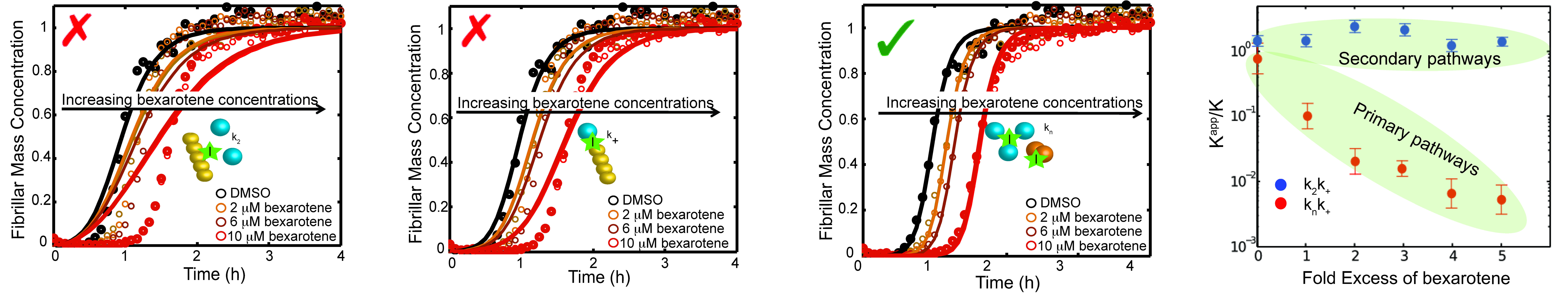
The conversion of the Abeta peptide into pathogenic aggregates is linked to the onset and progression of Alzheimer's disease. Although this observation has prompted an extensive search for therapeutic agents to modulate the concentration of Abeta or inhibit its aggregation, all clinical trials with these objectives have so far failed, at least in part because of a lack of understanding of the molecular mechanisms underlying the process of aggregation and its inhibition. To address this problem we describe a chemical kinetics approach for rational drug discovery, in which the effects of small molecules on the rates of specific microscopic steps in Abeta aggregation are analysed quantitatively. By applying this approach we report that bexarotene, an FDA-approved anti-cancer drug, targets selectively the primary nucleation step in Abeta aggregation, delays the formation of toxic species in neuroblastoma cells and completely suppresses Abeta deposition and its consequences in a C. elegans model of Abeta-mediated toxicity. These results suggest that the prevention of the primary nucleation of Abeta by compounds such as bexarotene could potentially reduce the risk of onset of Alzheimer's disease, and more generally that our strategy provides a general framework for the rational identification of a range of candidate drugs directed against neurodegenerative disorders.
Metainference: A Bayesian inference method for heterogeneous systems. Science Advances (2016).
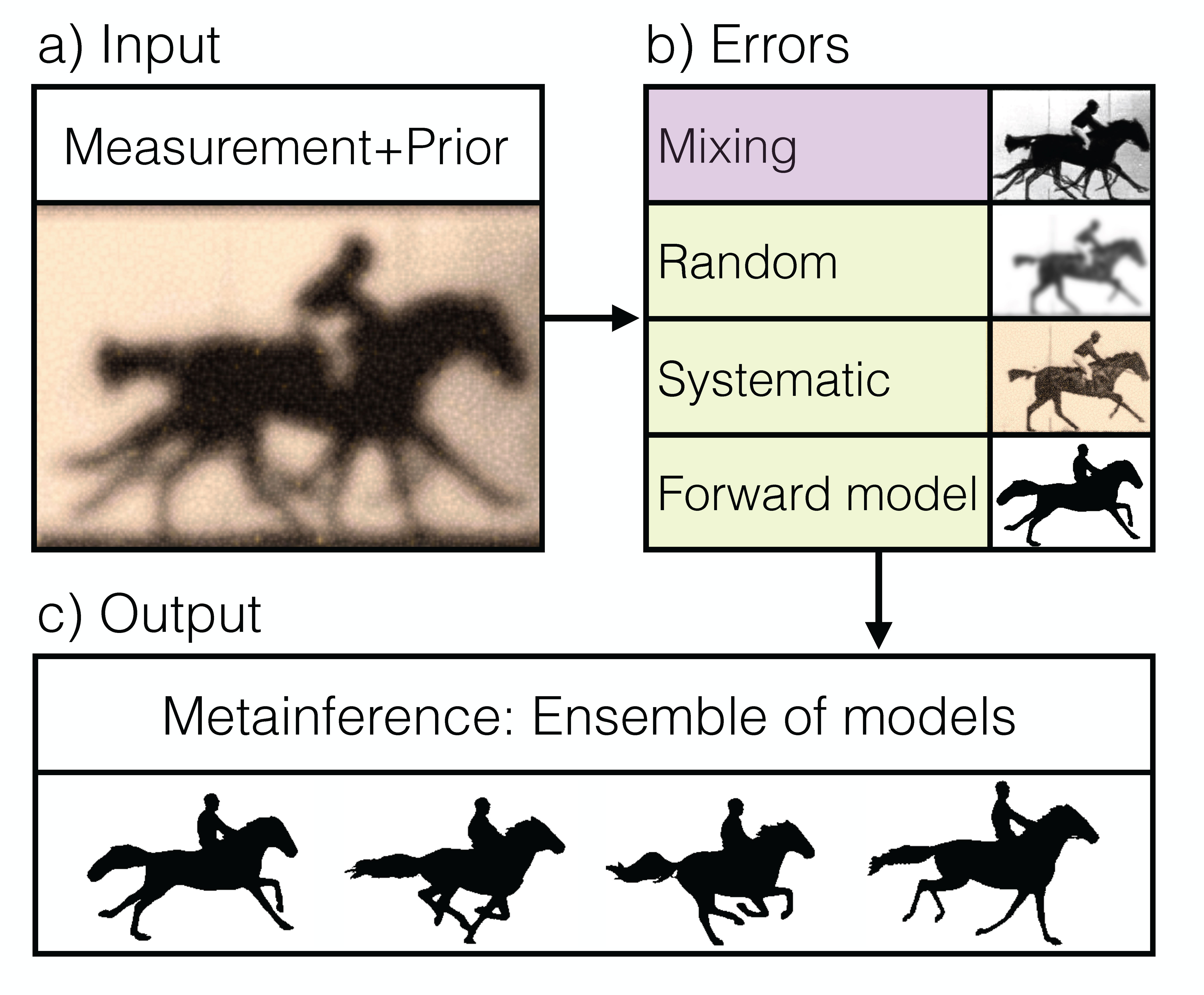
Modelling a complex system is almost invariably a challenging task. The incorporation of experimental observations can be used to improve the quality of a model, and thus to obtain better predictions about the behavior of the corresponding system. This approach, however, is affected by a variety of different errors, especially when a system populates simultaneously an ensemble of different states and experimental data are measured as averages over such states. To address this problem we present a Bayesian inference method, called 'metainference', that is able to deal with errors in experimental measurements as well as with experimental measurements averaged over multiple states. To achieve this goal, metainference models a finite sample of the distribution of models using a replica approach, in the spirit of the replica-averaging modelling based on the maximum entropy principle. To illustrate the method we present its application to a heterogeneous model system and to the determination of an ensemble of structures corresponding to the thermal fluctuations of a protein molecule. Metainference thus provides an approach to model complex systems with heterogeneous components and interconverting between different states by taking into account all possible sources of errors.
A rational design strategy for the selective activity enhancement of a molecular chaperone toward a target substrate. Biochemistry (2015)

Molecular chaperones facilitate the folding and assembly of proteins and inhibit their aberrant aggregation. They thus offer several opportunities for biomedical and biotechnological applications, as for example they can often prevent protein aggregation more effectively than other therapeutic molecules, including small molecules and antibodies. Here we present a method of designing molecular chaperones with enhanced activity against specific amyloidogenic substrates while leaving unaltered their functions toward other substrates. The method consists of grafting onto a molecular chaperone a peptide designed to bind specifically an epitope in the target substrate. We illustrate this strategy by describing Hsp70 variants with increased affinities for alpha-synuclein and Abeta42 but otherwise unaltered affinities for other substrates. These designed variants inhibit protein aggregation and disaggregate preformed fibrils significantly more effectively than wild-type Hsp70 indicating that the strategy presented here provides a possible route for tailoring rationally molecular chaperones for specific purposes.
Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA (2015)
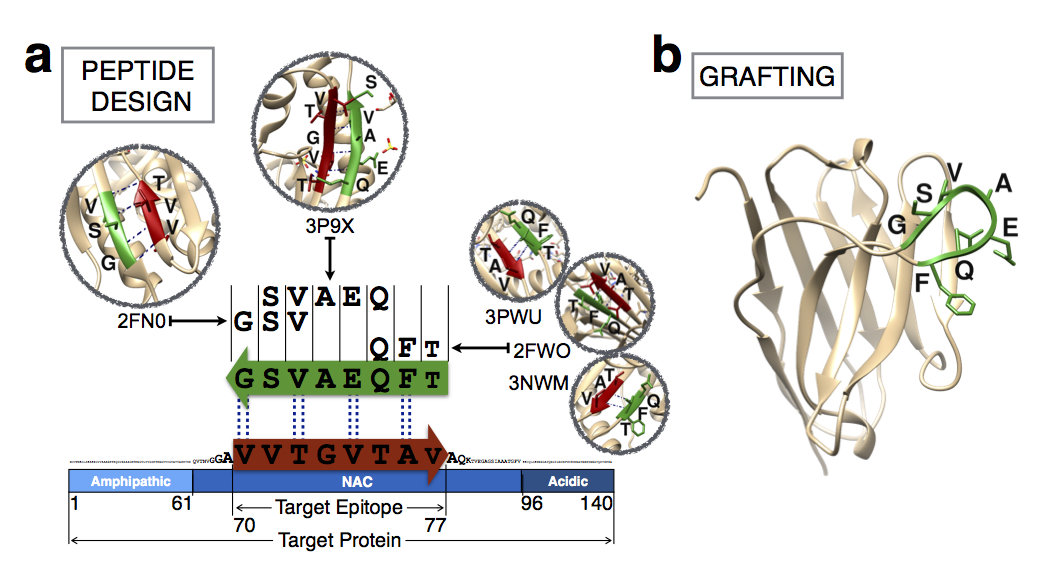
Antibodies are powerful tools in life sciences research, as well as in diagnostic and therapeutic applications, because of their ability to bind given molecules with high affinity and specificity. Using current methods, however, it is laborious and sometimes difficult to generate antibodies to target specific epitopes within a protein, in particular if these epitopes are not effective antigens. Here we present a method to rationally design antibodies to enable them to bind virtually any chosen disordered epitope in a protein. The procedure consists in the sequence-based design of one or more complementary peptides targeting a selected disordered epitope and the subsequent grafting of such peptides on an antibody scaffold. We illustrate the method by designing six single-domain antibodies to bind different epitopes within three disease-related intrinsically disordered proteins and peptides (alpha-synuclein, Abeta42, and IAPP). Our results show that all these designed antibodies bind their targets with good affinity and specificity. As an example of an application, we show that one of these antibodies inhibits the aggregation of alpha-synuclein at substoichiometric concentrations and that binding occurs at the selected epitope. Taken together, these results indicate that the design strategy that we propose makes it possible to obtain antibodies targeting given epitopes in disordered proteins or protein regions.
Structure of a low-population intermediate state in the release of an enzyme product. eLife (2015)
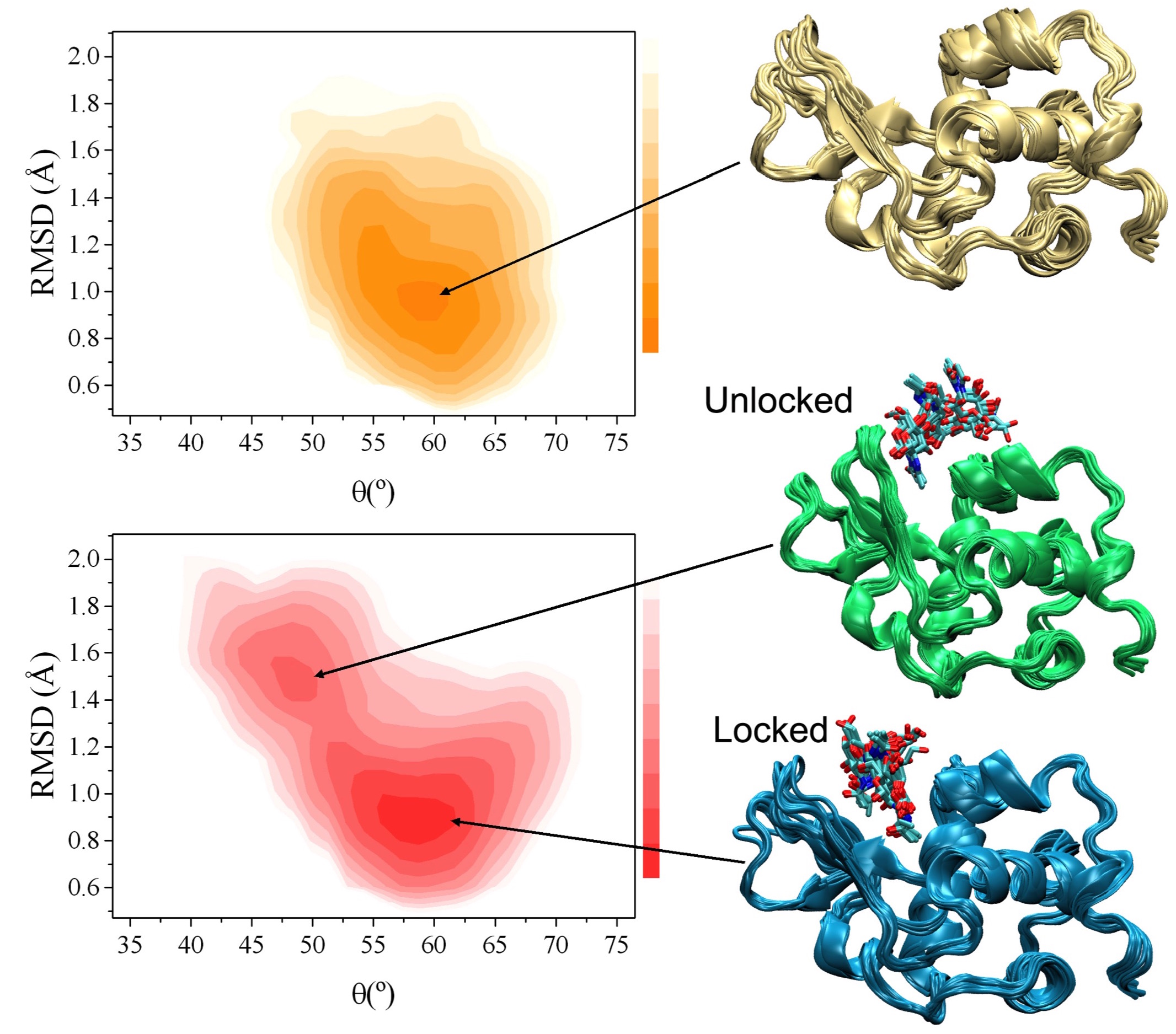
Enzymes can increase the rate of biomolecular reactions by several orders of magnitude. Although the steps of substrate capture and product release are essential in the enzymatic process, complete atomic-level descriptions of these steps are difficult to obtain because of the transient nature of the intermediate conformations, which makes them largely inaccessible to standard structure determination methods. We describe here the determination of the structure of a low-population intermediate in the product release process by human lysozyme through a combination of NMR spectroscopy and molecular dynamics simulations. We validate this structure by rationally designing two mutations, the first engineered to destabilise the intermediate and the second to stabilize it, thus slowing down or speeding up, respectively, product release. These results illustrate how product release by an enzyme can be facilitated by the presence of a metastable intermediate with transient weak interactions between the enzyme and the product.
A molecular chaperone breaks the catalytic cycle that generates toxic Abeta oligomers. Nat. Struct. Mol. Biol. (2015)
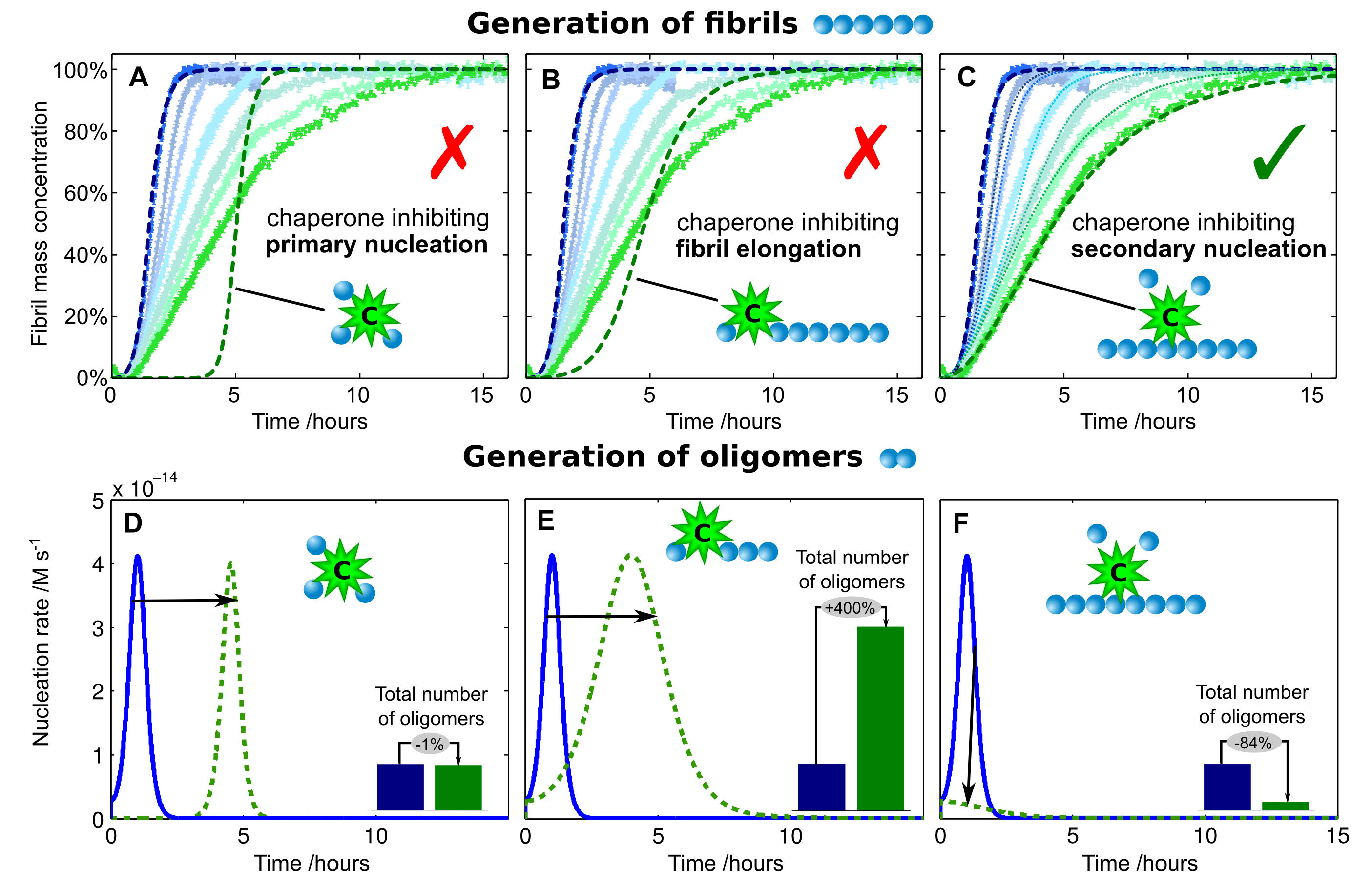
Alzheimer's disease is a highly debilitating and increasingly prevalent neurodegenerative disorder whose pathogenesis has been associated with the aggregation of the amyloid-beta peptide (Abeta42). Recent studies have revealed that the formation of highly neurotoxic oligomers during Abeta aggregation is strongly catalysed by the surfaces of larger aggregates. These findings indicate that a particularly effective way to limit Abeta42 toxicity would be through agents that inhibit this catalytic cycle. Here we show that a molecular chaperone, a Brichos domain, has the ability to specifically block the catalytic production of oligomers. We further demonstrate by a series of biophysical techniques that the Brichos domain achieves this inhibition in vitro by binding on the surface of the fibrils and redirecting the reactive flux from the monomeric to the fibrillar states via a pathway that involves a minimal formation of toxic oligomeric intermediates. We then verify by means of cytotoxicity and electrophysiology experiments using brain tissue that this mechanism also occurs in vivo. These results suggest that biological systems have evolved to achieve effective and efficient suppression of the toxic effects of protein misfolding and aggregation by targeting the individual microscopic pathways that create toxic oligomers, rather than perturbing the overall aggregation reaction.
Cyclophilin A catalyses proline isomerization by an electrostatic handle mechanism. Proc. Natl. Acad. Sci. USA 111, 10203-10208 (2014).
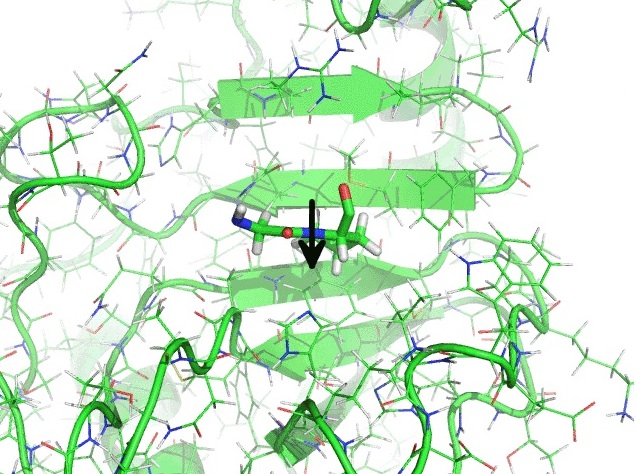
One of the most widespread molecular switches in biochemical pathways is based on the isomerization of the amino acid proline, a process that normally is facilitated by enzymes known as 'proline isomerases'. We show that cyclophilin A, one of the most common proline isomerases, acts by a simple mechanism, which we describe as an 'electrostatic handle'. In this mechanism, the enzyme creates an electrostatic environment in its catalytic site that rotates a peptide bond in the substrate by pulling the electric dipole associated with the carbonyl group preceding the peptide bond itself. Our results thus identify a specific mechanism by which electrostatics is exploited in enzyme catalysis.
Statistical mechanics of the denatured state of a protein using replica-averaged metadynamics. J. Am. Chem. Soc. 136, 8982-8991 (2014).

The characterization of denatured states of proteins is challenging because the lack of permanent structure in these states makes it difficult to apply to them standard methods of structural biology. In this work we use all-atom replica-averaged metadynamics (RAM) simulations with NMR chemical shift restraints to determine an ensemble of structures representing an acid-denatured state of the 86-residue protein ACBP. This approach has enabled us to reach convergence in the free energy landscape calculations, obtaining an ensemble of structures in relatively accurate agreement with independent experimental data used for validation. By observing at atomistic resolution the transient formation of native and non-native structures in this acid-denatured state of ACBP, we rationalize the effects of single-point mutations on the folding rate, stability, and transition-state structures of this protein, thus characterizing the role of the unfolded state in determining the folding process.
Widespread aggregation and neurodegenerative diseases are associated with supersaturated proteins. Cell Reports 5, 781-790 (2013).
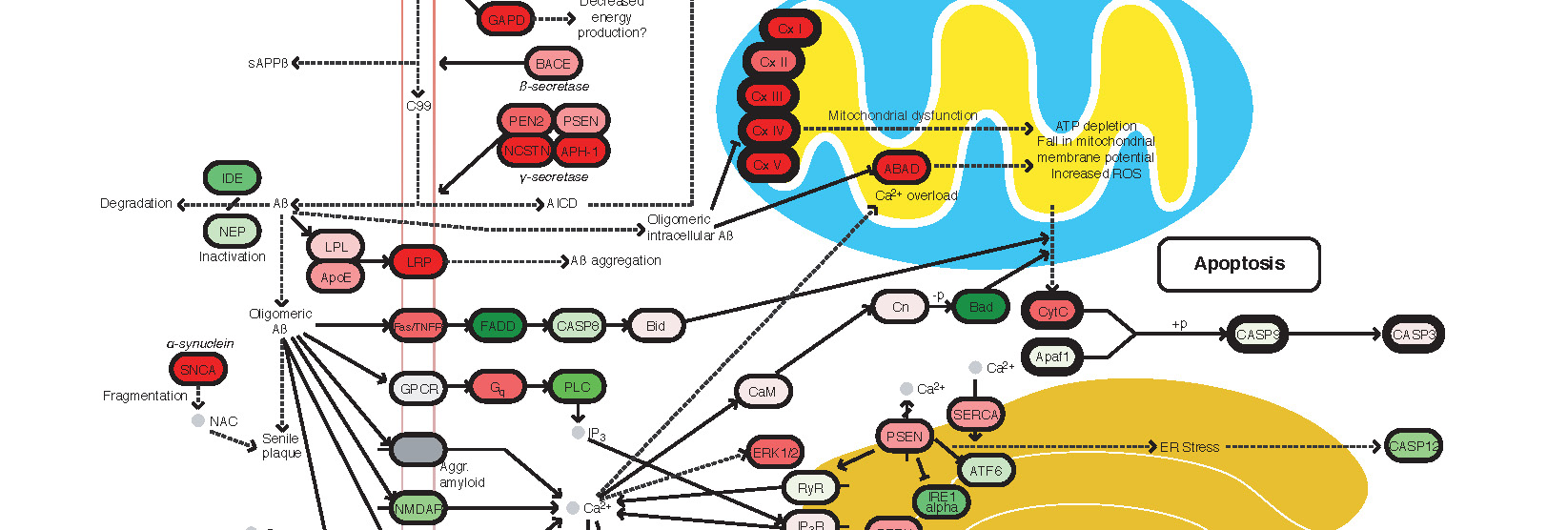
The maintenance of protein solubility is a fundamental aspect of cellular homeostasis because protein aggregation is associated with a wide variety of human diseases. Numerous proteins unrelated in sequence and structure, however, can misfold and aggregate, and widespread aggregation can occur in living systems under stress or aging. A crucial question in this context is why only certain proteins appear to aggregate readily in vivo, whereas others do not. We identify here the proteins most vulnerable to aggregation as those whose cellular concentrations are high relative to their solubilities. We find that these supersaturated proteins represent a metastable subproteome involved in pathological aggregation during stress and aging and are overrepresented in biochemical processes associated with neurodegenerative disorders. Consequently, such cellular processes become dysfunctional when the ability to keep intrinsically supersaturated proteins soluble is compromised. Thus, the simultaneous analysis of abundance and solubility can rationalize the diverse cellular pathologies linked to neurodegenerative diseases and aging.